

МЕХАНИЗМЫ МЕТОДИЧКА
.pdfВ обоих случаях скорость прямо пропорциональна константе скорости лимитирующей стадии продолжения цепи (ki), скорости зарождения цепи (r0) и обратно пропорциональна константе скорости обрыва цепи (kt). При наличии ингибиторов скорость обратно пропорциональна их концентрации.
При квадратичном обрыве цепи:
2r0 = rt = kt[X ∙]2 |
и |
[X ∙] = √ |
2r0 |
. |
|
kt |
|||||
|
|
|
|
Если в лимитирующей стадии продолжения цепи радикал Х· взаимодействует с веществом А, получаем:
r = ki[X ∙][A] = ki√2r0 [A] . kt
Как и в случае линейного обрыва, скорость прямо пропорциональна константе скорости лимитирующей стадии продолжения цепи, но от скорости зарождения цепи зависит в 0,5 степени, и в -0,5 степени — от константы скорости обрыва.
При использовании инициатора
r0 = k0[Inc]
выражение для общей скорости радикального процесса с квадратичным обрывом принимает вид:
|
2k0 |
[Inc] |
|
r = ki√ |
|
|
[A] . |
|
|
||
|
kt |
||
Зависимость вида кинетических уравнений радикально-цепных реакций от механизма зарождения, продолжения и обрыва цепи позволяет по экспериментальному уравнению скорости подобрать удовлетворяющий ему механизм реакции.
Отличительная особенность разветвленных цепных реакций состоит в том, что радикалы или молекулярные продукты, образующиеся на стадии продолжения цепи, могут начинать новые параллельные звенья процесса. Типичным примером являются процессы окисления, где значительная доля образующихся радикалов, а также гидропероксидов вступают в реакции либо с продуктами (или другими радикалами), либо с исходными веществами, давая при этом все новые радикалы.
111
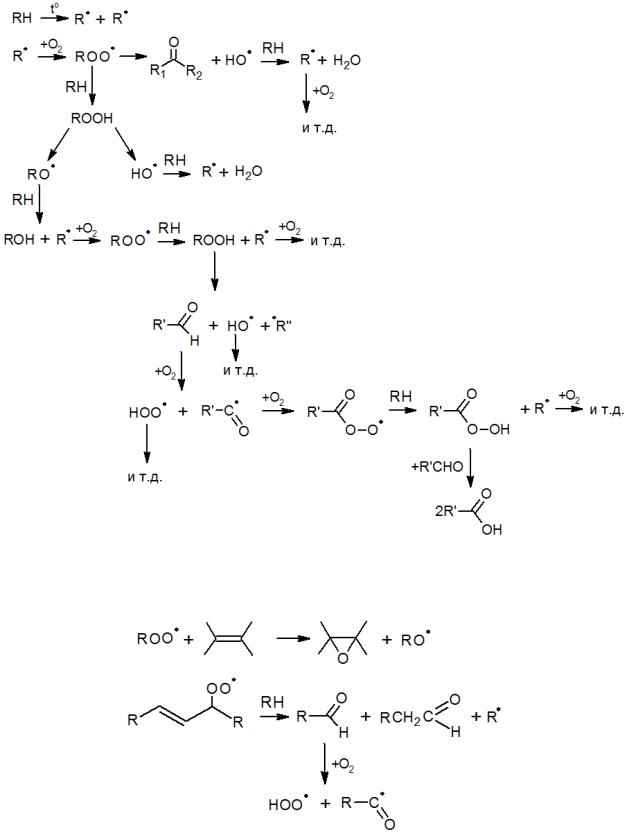
При окислении парафинов пути разветвления можно представить схематично следующим образом:
Если окислению подвергаются соединения с двойными связями, то имеют место и другие превращение, например:
112
Рассмотренные выше цепные и нецепные радикальные реакции также могут протекать и по ионным механизмам – как электрофильные и нуклеофильные реакции. Основным отличием радикальных реакций от гетеролитических (т.е. нуклеофильных и электрофильных, протекающих через стадию гетеролитического разрыва связи) следует признать меньшую избирательность в получении конкретных продуктов. Так, в зависимости от длины цепи в целевых продуктах реакции в соответствующих количествах будут находиться соединения, полученные при инициировании или обрыве цепей. В этом случае увеличение длины цепи приводит к лучшей избирательности методики получения целевого продукта. С другой стороны, для получения низкомолекулярных олигомеров или теломеров побочными продуктами будут более высокомолекулярные олигомеры. В этом случае следует создавать условия для уменьшения длины цепи радикальных реакций. Наилучшим выходом из такой ситуации является переход к более избирательному металлокомплексному катализу, идеалом которого являются ферментативные процессы.
5. РЕАКЦИИ, КАТАЛИЗИРУЕМЫЕ КОМПЛЕКСАМИ МЕТАЛЛОВ
В предыдущих разделах мы частично коснулись темы каталитических реакций, в которых катализаторами выступали, в основном, протон Н+ или ОН- анион. Однако существует множество других реакций, в которых катализаторами являются различные комплексы металлов. Эту область каталитических реакций выделяют отдельно, под названием «металлокомплексный катализ». Она сформировалась на основе развития ферментативного катализа, которым занимались биохимики. Поэтому в металлокомплексном катализе перенято много специфических терминов, употребляемых биохимиками.
Отличительной особенностью металлокомплексного катализа является высокая селективность катализируемых реакций, включая региоселективность и сохранение оптической активности. Такие результаты обеспечиваются тем, что реагенты взаимодействуют друг с другом в сфере комплекса металла. Кроме того, координация реагента у металла уменьшает энергию активации по сравнению с некоординированными молекулами.
113
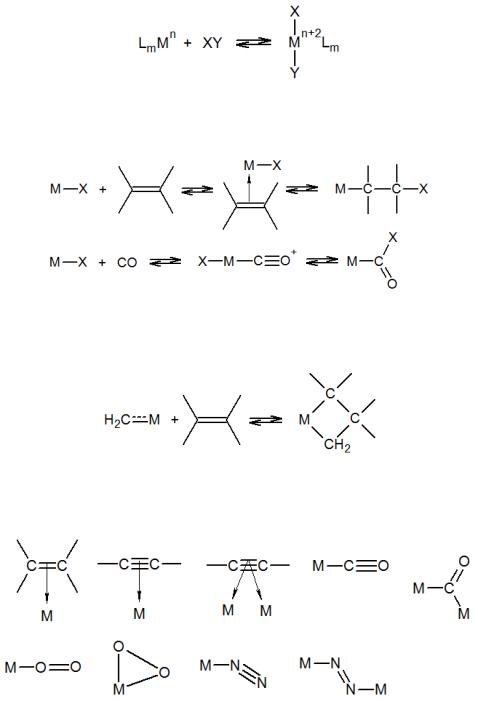
В металлокомплексном катализе выделяют несколько элементарных реакций, присущих только комплексным соединениям:
а) окислительное присоединение и обратное ему восстановительное элиминирование:
б) внедрение ненасыщенных соединений по связи М—X и обратная реакция – β-
элиминирование:
в) обратимое образование металлоцикла из ненасыщенных соединений и карбенов в координационной сфере металла:
г) активация молекул реагентов путем взаимодействия их - и -орбиталей с d- орбиталями металла при образовании различных комплексов:
Основные типы реакций, катализируемые комплексами металлов:
-гидрирование (гомогенное);
-димеризация, олигомеризация и полимеризация олефинов;
-изомеризация олефинов;
114
-метатезис олефинов;
-синтезы с участием оксида углерода;
-присоединение НХ к олефинам и ацетиленам;
-окисление углеводородов в карбонильные соединения и эпоксиды;
-активация алканов в растворах.
5.1. Гомогенное гидрирование
Гидрирование – взаимодействие органических веществ с молекулярным водородом, которое может приводить к насыщению кратных связей, восстановлению органических соединений, а также деструкции углерод– углеродных связей. Практически все реакции гидрирования обратимые.
Процесс гидрирования всегда проходит с выделением тепла и очевидно, что равновесие будет смещаться в сторону продуктов при понижении температуры. Смещению равновесия в сторону продуктов реакции способствует также повышенное давление, поскольку реакция гидрирования протекает с поглощением водорода (т.е. с уменьшением объема реакционной системы).
По активности в реакции гидрирования различные классы органических соединений располагаются в следующем ряду:
алкены > алкины > ароматические углеводороды;
альдегиды >кетоны > нитрилы > эфиры карбоновых кислот > карбоновые кислоты.
Реакционная способность олефинов зависит от заместителей атомов углерода при двойной связи: чем больше заместителей, тем меньше скорость реакции вследствие пространственного перекрывания двойной связи. Соответственно, легче всего гидрируется этилен, далее реакционная способность падает в соответствии со следующим рядом:
CH2=CH2 > RCH=CH2 > RCH=CHR > R2C=CH2 > R2C=CHR > R2C=CR2
Скорость гидрирования диенов и полиенов выше, чем олефинов, и также зависит от строения углеводородного скелета. Реакция протекает последовательно с уменьшением количества двойных связей в молекуле; полное глубокое гидрирование приводит к образованию парафинов.
Карбоновые кислоты восстанавливаются водородом последовательно через образование альдегидов, спиртов, и в конечном итоге образуются углеводороды.
115
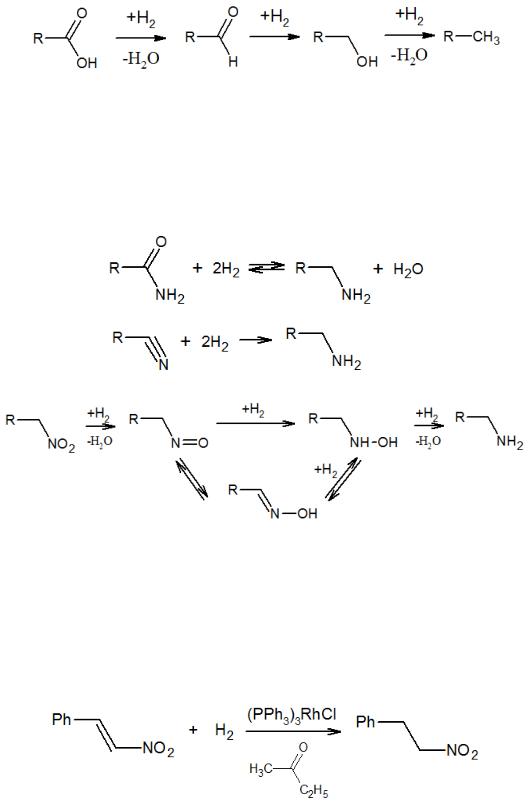
Обычно такие реакции используют для получения спиртов, поскольку альдегиды реагируют с водородом намного быстрее кислот и в данном случае их выделить не удается.
Целью гидрирования различных азотсодержащих соединений обычно является получение аминов. Для этого реакции подвергают амиды кислот, нитрилы, нитросоединения. Последние реагируют через ряд промежуточных стадий, включающих образование нитрозосоединений и гидроксиламинов. Алифатические нитрозосоединения могут изомеризоваться в оксимы.
Проведение глубокого гидрирования иногда сопровождается образованием нежелательных продуктов гидрогенолиза, то есть разрыва С–С-связи под действием водорода. Обычно, реакцию останавливают в определенный момент, препятствуя расходованию целевого продукта на дальнейшие превращения.
В случае гидрирования функционально замещенных алкенов группы COOR, NO2, CN, C(O)R не затрагиваются, например:
116
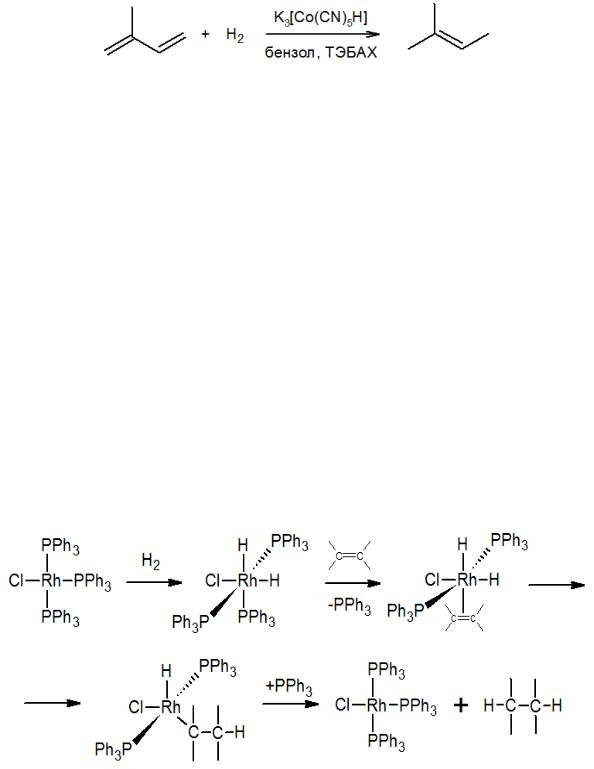
В данной реакции в качестве растворителя использован кетон, который, как и группа NO2, в этих условиях не восстанавливается.
Гомогенное гидрирование можно проводить в двухфазной системе. При этом в качестве катализатора используется соль K3[Co(CN)5H]. Эта соль с помощью катализатора межфазного переноса (здесь, триэтилбензиламмоний хлорид) переводится в органическую фазу, где и катализирует гидрирование.
Наиболее известными и эффективными катализаторами гомогенного гидрирования являются комплексы хлоридов родия (I) и рутения (III) с трифенилфосфином – трис-(трифенилфосфин)-родийхлорид [(C6H5)3P]3RhCl (катализатор Дж. Уилкинсона) и гидрохлорид трис-(трифенилфосфин)-рутения [(C6H5)3P]3RuHCl. С точки зрения промышленного применения гомогенный катализ уступает гетерогенному, в основном, ввиду сложностей, связанных с отделением катализатора от реакционной массы. Тем не менее, благодаря достаточно высокой селективности гидрирования, гомогенный катализ применяют для восстановления сложных полифункциональных соединений. Чаще всего применяют родиевый комплекс – катализатор Дж. Уилкинсона. Реакцию проводят при комнатной температуре и атмосферном давлении. Механизм гомогенного гидрирования включает в себя следующие этапы:
-координацию молекулы водорода с атомом металла и последующее окислительное присоединение;
-замещение одного из фосфиновых лигандов на алкен, образование π-комплекса;
-внедрение алкена по связи металл–водород;
-восстановительное элиминирование продукта гидрирования.
117
Лимитирующей стадией является атака алкена на дигидридный комплекс. Несмотря на то, что присоединение атомов водорода происходит не одновременно, стереохимический результат можно описать как син-присоединение (т.е. присоединение идет с только одной стороны олефина, а при гидрировании ацетиленов – с образованием цис-продуктов). Восстановленный катализатор вновь повторяет цикл превращений. Активность катализатора характеризуется числом таких циклов в секунду, называемым числом оборотов (Turnover number) или
частотой оборотов TOF (Тurnover frequency).
Для обычных гомогенных катализаторов это число составляет 10−7-10−2с− 1, тогда как для ферментов до 106 с−1. Сопоставление этих величин показывает, насколько еще несовершенны искусственно созданные катализаторы.
5.2. Полимеризация, олигомеризация и димеризация олефинов
Полимеризация – процесс образования высокомолекулярного вещества (полимера) путем многократного присоединения молекул низкомолекулярного вещества (мономера, олигомера) к активным центрам в растущей молекуле полимера. Реакции полимеризации подразделяют на ступенчатую и цепную полимеризацию. Ступенчатая полимеризация протекает таким образом, что образующиеся на каждой стадии олигомеры могут быть выделены. Как правило, таким способом получают ди-, три- и тетрамеры. Ступенчатая полимеризация значительно меньше распространена, чем цепная. Цепная полимеризация приводит к образованию высокомолекулярного соединения, независимо от механизма реакции — радикально–цепной или металлокомплексный катализ. В отличие от радикальной или ионной (катионной) полимеризации, рост цепи в каталитической реакции происходит в координационной сфере металла. Такой процесс осуществляется на моногидридных комплексах металлов: при последовательном внедрении молекул олефина по связи М–Н, и далее по связи М–С:
118
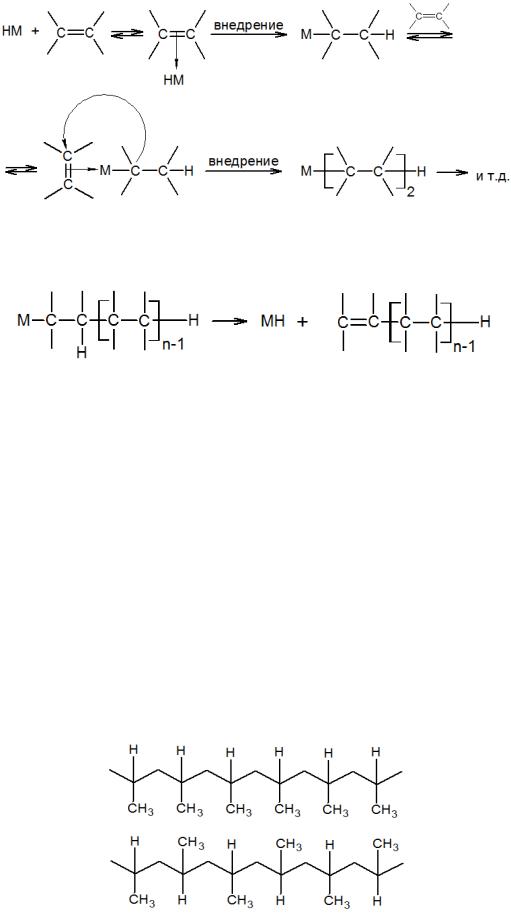
В реакции внедрения вступают только монозамещенные олефины с концевой двойной связью. Рост полимерной цепи заканчивается реакцией элиминирования с регенерацией исходного гидрида металла:
Для такого процесса необходимы комплексы металлов переменной валентности, которые способны образовывать металл–алкильные производные М–R. Для металлов переменной валентности такие соединения, как правило, малоустойчивы, поэтому для их образования необходимо присутствие достаточно стабильных металл–алкильных соединений, которые способны к реакциям, регенерирующим первоначальный комплекс с М–С-связью. Такими со-катализаторами могут быть алкильные производные Al, Li, Mo, Sn и другие. Так, при взаимодействии Аl(С2Н5)3 с (η-C5H5)2TiCl2 в углеводородных растворителях образуются катализаторы полимеризации этилена и α-олефинов (катализаторы Циглера–Натты). Полимеризация пропилена и других олефинов происходит с образованием стереорегулярных полимеров, то есть таких полимеров, в которых звенья располагаются по отношению друг к другу определенным образом, и такая расстановка периодически повторяется по всей длине молекулы. Например, стереорегулярный полипропилен может выглядеть следующим образом:
119
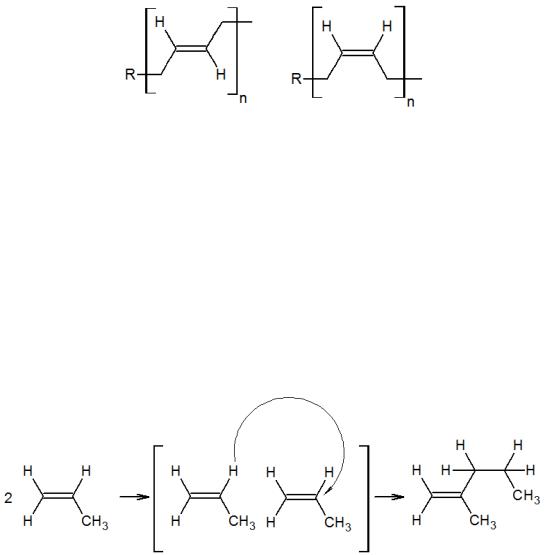
Бинарные системы СоСl2-АlСl(С2Н5)2, VСl3(тетрагидрофуран)3–АlСl(С2Н5)2 используют для получения стереорегулярных бутадиеновых каучуков. В присутствии первого комплекса образуется полимер, содержащий 93–98% звеньев структуры 1,4-цис. Второй катализатор приводит к получению полимера с 99 % 1,4- транс-структурой.
Олигомеризация олефинов – процесс образования низкомолекулярного вещества (олигомера) путём присоединения определенного количества молекул мономера к активным центрам в растущей молекуле. Процесс фактически повторяет элементарные стадии полимеризации, но в заданный момент реакцию останавливают.
Механизм реакции олигомеризации идентичен механизму полимеризации. Однако в отличие от высокомолекулярных полимеров, для олигомеров и их чистоты большую роль играют концевые группы олигомерной цепи, в частности, при получении димеров. Образование линейных олигомеров связано с переносом водорода от одного участника реакции к другому, например:
5.3. Изомеризация олефинов
Изомеризация – реакция превращения молекулы в ее изомер – другую молекулу, имеющую одинаковую молекулярную, но разную структурную формулу. Различают изомеризацию, связанную с миграцией двойной связи и скелетную изомеризацию.
120