

Современные проблемы и методы биотехнологии
.pdfГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
Чтобы колонка не «садилась», необходимо производить набивку колонки при давлении в 3–4 раза выше ее рабочего давления (т.е. 300–400 кгс/см2). Для хорошей колонки N = 30 000–100 000 т.т./м.
Детекторы. Детектор является преобразователем концентрации анализируемого вещества, растворенного в подвижной фазе, в электрический сигнал.
В современном жидкостном хроматографе для детектирования компонентов пробы может быть использовано любое физико-химическое свойство подвижной фазы (поглощение света, излучение света, электропроводность, показатель преломления и т.д.), которое изменяется при наличии в ней молекул разделяемых соединений (табл. 6.1). Из существующих 50 физикохимических методов детектирования в настоящее время активно используется 5–6.
Таблица 6.1
Основные типы детекторов, используемые в ВЭЖК
|
|
|
Ориентиро- |
|
|
Детекторы |
Измеряемые свойства подвижной фазы |
вочная чувст- |
Селек- |
|
вительность, |
тивность |
||
|
|
|
мг |
|
1. |
Фильтровой фо- |
Оптическая плотность на определенной |
10-10 |
Высокая |
тометрический |
длине волны, пропускаемой фильтром |
|
|
|
2. |
Спектрофото- |
Оптическая плотность на выбранной |
10-9 |
Высокая |
метрический |
длине волны монохроматора |
|
|
|
3. |
Рефрактометри- |
Разность показателей преломления рас- |
10-6 |
Низкая |
ческий |
творителя и раствора с пробой |
|
|
|
4. |
Флуорометриче- |
Интенсивность излучения молекул про- |
10-11 |
Очень |
ский |
бы в элюенте |
|
высокая |
|
5. |
Амперометриче- |
Ток окисления и восстановления элек- |
10-9–10 -11 |
Очень |
ский |
трохимически активных веществ |
|
высокая |
|
Кондуктометриче- |
Электропроводность пробы в элюенте |
10-6 |
Низкая |
|
ский |
|
|
|
|
Наибольшей сложностью при конструировании хроматографических детекторов было сочетание малых объемов ячеек (0,1–10 мкл, менее 10 % от объема неудерживаемого пика) с высокой чувствительностью (ввиду малого объема и низкой концентрации пробы). Иллюстрацией достигнутых успехов является то, что чувствительность фотоколориметров и спектрофотометров составляет 10–2 ед. оптической плотности, в то время как чувствительность фотометрических детекторов – 10–5 ед. оптической плотности, т.е. в 1 000 раз выше.
Чувствительность − это важнейшая характеристика детектора. Лучше всего оценивать этот параметр по физической величине. Если определять чувствительность через двойную амплитуду шума нулевой линии, шум выражать в физических единицах, то чувствительность фотометрического де-
Современные проблемы и методы биотехнологии. Учеб. пособие |
321 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
тектора будет выражаться в единицах оптической плотности, рефрактометрического − в единицах показателя преломления, вольт-амперометрического − в амперах, кондуктометрического − в сименсах. Для химика, очевидно, интереснее определять чувствительность в минимальном количестве определяемого вещества.
Чувствительность детектора может быть примерно одинаковой ко всем компонентам пробы (рефрактометр и кондуктометр), а может быть совершенно разной даже для близких соединений. В первом случае говорят о неселективном детектировании. Это значит, что измеряется физическое свойство, присущее и пробе и растворителю (показатель преломления, электропроводность), их разность. Во втором случае− селективное детектирование. Это значит, что измеряется физическое свойство, присущее только молекулам пробы, например, способность флуоресцировать или поглощать свет. Селективное детектирование, с одной стороны, позволяет повысить чувствительность определения или исключить те вещества, которые определять не нужно (предельные углеводороды при определении ароматики), с другой стороны, допускает возможность не обнаружить нужных нам компонентов (тех же предельных углеводородов в нефти). Поэтому при исследовании общего состава объекта лучше использовать неселективный детектор типа рефрактометра, а при определении концентрации отдельных компонентов в сложной смеси – селективные детекторы.
Подвижные фазы (ПФ). Элюирующая сила и эффективность
Подвижная фаза в ЖХ выполняет двоякую функцию: с одной стороны, она обеспечивает перенос десорбированных молекул по колонке. Для этого химические свойства подвижной фазы не играют существенной роли, более важны их физические свойства: вязкость, летучесть. С другой стороны, подвижная фаза в ЖХ играет активную химическую, по существу, роль. Молекулы подвижной фазы взаимодействуют с другими компонентами системы: молекулами разделяемых веществ и молекулами неподвижной фазы. Поэтому вторая и более важная функция подвижной фазы сводится к регулированию констант равновесия, к регулированию удерживания. Возможности регулирования удерживания с помощью подвижной фазы необычайно велики. Нередко заменой одного растворителя на другой, можно изменить коэффициент емкости К' в 1 000–10 000 раз, однако для практической хроматографии пригоден лишь довольно узкий диапазон величин значение К' − примерно 1–20. Слишком малые значения К' непригодны, т.к. в этой области резко возрастает вероятность взаимного перекрытия пиков. Наоборот, если К' слишком велико, для разделения требуется слишком много времени, к то му же увеличивается риск не обнаружить более прочно сорбирующиеся компоненты смеси.
Таким образом, для решения каждой конкретной задачи состав как подвижной, так и неподвижной фазы должен быть тщательно подобран по физическим и химическим свойствам ее компонентов. Основными характеристиками подвижных фаз являются их элюирующая сила и селективность.
Современные проблемы и методы биотехнологии. Учеб. пособие |
322 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
Один из элюентов способен смыть с колонки лишь слабосвязанные сорбаты, другие же вызывают десорбцию почти любых молекул. Молекулы подвижной фазы могут взаимодействовать с молекулами разделяемых веществ. При этом образуются ассоциаты, которые взаимодействуют с неподвижной фазой, сила этого взаимодействия отличается от силы взаимодействия молекул пробы. В результате сорбция может стать более или менее прочной. С другой стороны, молекулы подвижной фазы могут конкурировать на поверхности сорбента с молекулами разделяемых соединений, вытесняя последние с активных центров и способствуя смещению равновесия в сторону десорбции. Для характеристики влияния подвижной фазы на удерживание используют понятие элюирующей силы.
Элюирующая сила подвижной фазы− это ее свойство вступать в такие межмолекулярные взаимодействия с компонентами системы, которые способствуют десорбции разделяемых соединений, более быстрому перемещению хроматографических зон.
Один подходящий растворитель подобрать трудно. Зато смесь растворителей очень увеличивает гибкость ВЭЖХ. Принцип составления таких смесей прост. Необходимо взять два индивидуальных растворителя, один из которых имеет заведомо недостаточную элюирующую силу (А1, А2), другой− заведомо избыточную (В1). Из этих двух растворителей можно приготовить множество различных подвижных фаз. Часть из них будет обязательно обладать подходящей элюирующей силой (А2, В1). Сила растворителя является экспериментально определяемым интегральным параметром. Когда мы говорили, что растворитель имеет подходящую силу, под этим имеется ввиду, что на данном сорбенте данный сорбат будет иметь приемлемое значение К'.
В то же время ясно, что для разделения уже двух соединений подойдет не любая из подвижных фаз, имеющих необходимую силу. Вещества могут иметь хорошее удерживание, но часть пиков перекрывается. В таких случаях мы говорим, что селективность системы недостаточна. Селективность определяется селективностью подвижной фазы, что связано со специфическими взаимодействиями с сорбатами. Селективность, как и элюирующая сила бинарной подвижной фазы, определяется в первую очередь природой более сильного ее компонента. В составе почти любой ПФ можно найти компонент сорбционно менее активный, выполняющий преимущественно транспортную функцию (растворитель А), и сорбционно активный, который служит для регулирования равновесия (растворитель В). Роль одного и того же компонента в различных подвижных фазах и в зависимости от характера НФ, может быть различной. Например, в ПФ гексан-хлороформ последнее соединение выступает в качестве растворителя В (активного), а в системе хлороформ− мет а- нол оно же играет роль растворителя А (транспортного). С целью повышения селективности часто используют подвижные фазы более сложного состава, чем бинарные. Во многих случаях это приводит к улучшению разделения.
Растворители. Растворители должны быть очищены от механических примесей. Примеси вызывают плохую работу клапанов, способствуют износу плунжеров и уплотнений, забивают входные фильтры колонок. Поэтому рас-
Современные проблемы и методы биотехнологии. Учеб. пособие |
323 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
творители нужно фильтровать через материал с порами 0,5 мкм, при отборе растворителя из резервуара тоже нужен фильтр. Если температура кипения растворителя низкая (ниже 60 °С) − ацетон, метиленхлорид, пентан, эфир и т.д., то при всасывании могут образоваться паровые пузыри, препятствующие работе клапанов. У менее летучих растворителей больше вязкость, что ведет к увеличению сопротивления колонки и к повышению давления на входе при сохранении расхода. Вязкость более 1,5 сП не желательна.
В зависимости от метода детектирования, к растворителям предъявляются определенные требования (табл. 6.2). При УФ, работающем при 190–220 нм, применимы вода, ацетонитрил, метанол, этанол, гексан, гептан, циклогексан и т.д. Поглощают обычно примеси, поэтому надо использовать не ХЧ, а «для ЖХ» или «для спектроскопии».
Таблица 6.2
Элюотропный ряд − перечень растворителей в порядке возрастания элюирующей способности
Растворитель |
Элюирующая сила на силикагеле |
Гексан |
0,01 |
Бензол |
0,10 |
Бутилхлорид |
0,20 |
Хлороформ |
0,26 |
Метиленхлорид |
0,32 |
ИП эфир |
0,34 |
Этилацетат |
0,38 |
Тетрагидрофура |
0,44 |
Ацетонитрил |
0,50 |
Метанол |
0,70 |
Перечень наиболее часто используемых растворителей:
–Вода (лучше бидистиллят, еще лучше деионизованная и очищенная от органики).
–Спирты (метанол, этанол, изопропанол)− годятся и ХЧ. Для обраще- но-фазовой хроматографии лучше использовать метанол – у него меньше всего вязкость (0,54 сп), для НФ− изопропанол, неограниченно смешива ю- щийся с углеводородами.
–Ацетонитрил − удобен для ОФХ (низкая вязкость 0,34 сп) и хорошая растворимость.
–Ароматика − поглощает в УФ и токсична.
–Алканы − гексан в НФ (недостаточно растворяет).
Специфические модификаторы ПФ вводятся для изменения свойств сорбента в лучшую сторону, улучшения разделения. Уксусная кислота 0,5–2,0 % − используется для аммоний-ацетатных буферных растворов, поглощает ниже 230 нм. Фосфорная кислота− в ОФХ д ля буферных растворов или для создания кислой среды. Аммиак− буферы в ОФХ и ИОХ. Алки л- сульфаты натрия (от С2 до С12)− добавки для перевода в ион -парный ре-
Современные проблемы и методы биотехнологии. Учеб. пособие |
324 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
жим, рН 2–5. Величина удерживания оснований растет. Тетра и триалкиламмоний − ион-парный вариант определения кислот (с ростом концентрации ТБА растет удерживание анионов). Аммиак− буферы в ОФХ и ИОХ.
Алкилсульфаты натрия (от С2 до С12)− добавки для перевода в ион -парный режим, рН 2–5.
Выбор условий разделения. Распределение молекул сорбатов между неподвижной и подвижной фазами определяется, в первую очередь, сравнительным сродством этих молекул к полярным или неполярным растворителям (или поверхностям). Это сродство является главным фактором, влияющим на величины удерживания, выбор неподвижной и подвижной фаз. Так, при использовании неполярных НФ (обращенно-фазная хроматография) удерживание ряда сорбатов уменьшается с увеличением их полярности, а удерживание данного сорбата на данной НФ уменьшается с уменьшением полярности ПФ, т.е. при добавлении органических растворителей. В противоположность этому, при хроматографии на полярных сорбентах (нормаль- но-фазная хроматография) удерживание растет с ростом полярности сорбатов и уменьшением полярности ПФ. То есть «подобное притягивается подобным».
Ряд правил, которые надо помнить при выборе условий разделения:
1.Адсорбционная хроматографическая система обладает удерживающей способностью только в тех случаях, когда условная полярность ПФ и НФ различается в достаточной степени. Величины удерживания тем больше, чем больше разница в полярности, химической природе фаз.
2.Приемлемые величины удерживания достигаемы только в таких случаях, когда полярность сорбата является промежуточной по сравнению с полярностью ПФ и НФ. Поэтому основной способ влияния на удерживание на данном сорбенте − изменение полярности подвижной фазы.
3.При хроматографии на силикагеле удваивание концентрации полярного компонента в ПФ ведет к уменьшению удерживания в 2,0–2,5 раза. При хроматографии на алкилсиликагелях (С2, С8, С18) уменьшение концентрации растворителя в 1, 2 раза приводит к 2,0–2,5 кратному уменьшению удерживания сильно сорбирующихся веществ (К' > 10) и 1,2–1,5 кратному уменьшению для слабо сорбирующихся (К' < 2).
4.При хроматографии ионогенных соединений, если рН среды такое, что допускается одновременное существование сорбата в нейтральной и ионизированных формах, степень ионизации в сорбированном и десорбированном состоянии различна. Следствие− снижение эффективности разделения, нарушение формы пика. Поэтому надо использовать ПФ на основе буферных растворов. Без этого трудно добиваться воспроизводимости и качественного
разделения. В общем с понижением рН TR оснований падет, а кислот растет.
Выбор хроматографической системы. Под термином «тип хромато-
графической системы» понимают сочетание: сорбента того или иного химического класса, подвижной фазы на основании водных или органических растворителей, характеризующейся интервалом рН, наличием специфических добавок кислого, основного или поверхностно-активного характера и т.д. Вы-
Современные проблемы и методы биотехнологии. Учеб. пособие |
325 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.2. Газожидкостнаяи высокоэффективная жидкостнаяхроматография для опр-я кол-ых и кач-ххаракт-кцелевых прод-в биотехнологии
бор типа системы основывается в первую очередь по оценке полярности и кислотно-основных свойств компонентов изучаемой системы. Условная оценка полярности анализируемых веществ, понимаемой в данном случае как относительное количество полярных и неполярных структурных фрагментов, представленных в их молекулах, может быть основана на шкале упрощенного критерия гидрофобности Н. Этот критерий рассчитывается по формуле
Н = nH −4 |
nf |
, |
(6.30) |
где nH − суммарное число атомов углерода и галогенов; nf − число функциональных групп в молекуле сорбата.
Чем больше Н, тем меньше полярность, выше гидрофобность (водоотталкивание). При хроматографии неполярных сорбатов средней полярности очень часто оказываются пригодными как нормально-, так и обращеннофазные системы. Для сорбатов с низкой (ароматика) или высокой (органические кислоты, спирты) полярностью пригодны ОФХ. Силикагель (НФХ) селективнее при разделении изомеров (о, м, п), а также сорбатов, различающихся по функциональности (производные бензола, фенола и т.д.). При разделении гомологов (пентан− декан), спиртов , кислот лучше применять алкилсиликагели (ОФХ). Если в анализируемой смеси предполагаются компоненты разной химической природы: витамины, лекарства, наркотики, токсины, гормоны начинать разработку метода лучше с ОФХ.
Выбор элюирующей силы и селективности подвижной фазы
При работе в ОФХ выбор элюирующей силы сводится к выбору концентрации органического компонента подвижной фазы. В первичном случае может быть использован подход на зависимости удерживания от Н. Если известны принадлежность сорбата к определенному классу веществ и модель удерживания этого класса в ОФХ, то задача упрощается. Для такого расчета концентрации элюента есть уравнение:
lgCx = |
1,4 |
−(1,4 −lgC)(lg Kx′ |
+0,7) |
, |
(6.31) |
|
|
|
lg Kx′ +0,7 |
|
|||
|
|
|
|
|
|
|
где K′ − коэффициент емкости при концентрации растворителя С, Сх − концентрация растворителя, необходимая для получения желаемого K′х.
В НФХ длянаименее полярных фаз (15 > H > 10) используют гексан:ИПС (95:5) или гексан:хлороформ (1:1). Для среднеполярных (10 > H > 5) – гексанИПС (80:20) или хлороформ:метанол (95:5). Для полярных (5 > H > 0)– хлороформ:метанол (80:20).
Современные проблемы и методы биотехнологии. Учеб. пособие |
326 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.3. Масс-спектрометриявбиотехнологии
Масс-спектрометрия − это физико-химический метод измерения отношения массы ионов к их заряду. Приборы, которые используются в этом методе, называются масс-спектрометрами или масс-спектрометрическими детекторами, они имеют дело с материальным веществом, состоящим, как известно, из мельчайших частиц− молекул и атомов. Масс-спектрометры устанавливают молекулярную массу вещества, ее атомарный и изотопный состав, а также пространственную структуру расположения атомов.
Как аналитический метод масс-спектрометрия обладает исключительно высокой чувствительностью и позволяет обнаруживать следовые количества органического вещества в больших объемах газов и жидкостей, а также в биологических системах. С помощью масс-спектрометрии можно изучать превращения вещества в процессе химической реакции, что существенно для установления ее механизмов. Значительное отличие масс-спектрометрии от других аналитических физико-химических методов заключается в том, что оптические, рентгеновские и некоторые другие методы детектируют излучение или поглощение энергии молекулами или атомами (например, такие методы, как ИК-, УФ-, КР- и ЯМР), а масс-спектрометрия имеет дело с самими частицами вещества, и в отличие от вышеуказанных методов, является деструктивным методом анализа, т.е. из образующихся при разрушении молекулы ионов исходная молекула регенерироваться не может. Метод массспектрометрии основан на ионизации молекул, разделении ионов в газовой фазе, которое происходит в зависимости от соотношения их массы и заряда, и регистрации разделенных ионов.
Для представления масс-спектральных данных часто пользуются графиками, называемыми масс-спектрами. Масс-спектр − это распределение заряженных частиц по их интенсивностям в соответствии с отношениями их масс к зарядам. Следовательно, первое, что надо сделать для того, чтобы получить масс-спектр, перевести нейтральные молекулы и атомы, составляющие любое органическое или неорганическое вещество, в заряженные частицы − ионы. Этот процесс называется ионизацией и осуществляется различными способами.
Современные проблемы и методы биотехнологии. Учеб. пособие |
327 |
ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.3. Масс-спектрометрия в биотехнологии
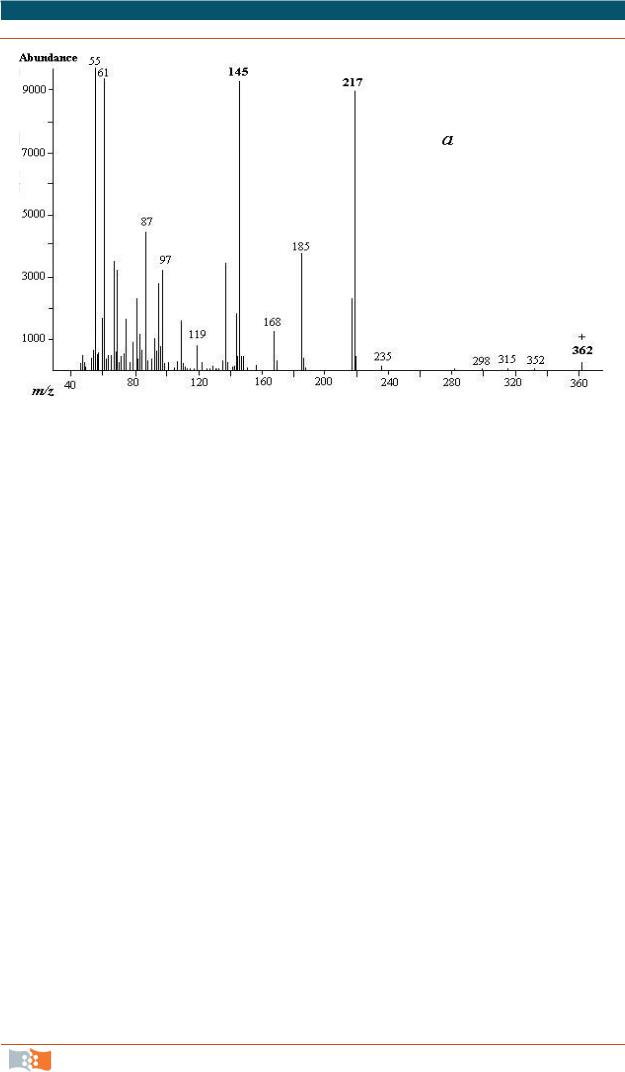
Рис. 6.14. Масс-спектр диметисульфоксидного производного пальмитолеиновой кислоты из Ralstonia eutropha. Молекулярная масса компонента составляет 362, а идентифицирующими ионами положения двойной связи в молекуле являются 145 и 217 m/z [11]
Следовательно масс-спектрометрия (масс-спектральный анализ) – метод анализа вещества путем определения массы (чаще отношения массы к заряду m/z) и относительного количества ионов, получаемых при ионизации исследуемого вещества или уже присутствующих в изучаемой смеси. Совокупность значений m/z и относительных величин токов этих ионов, представленная в виде графика или таблицы, называется масс-спектром вещества. В качестве иллюстрации приведен масс-спектр дисульфидного производного метилового эфира пальмитолеиновой кислоты, выделенной из бактерий Ralstonia eutropha (рис. 6.14).
Начало развитию масс-спектрометрии положено опытами Дж. Томсона (1910), исследовавшего пучки заряженных частиц, разделение которых по массам производилось с помощью электрического и магнитных полей, а спектр регистрировался на фотопластинки. Первый масс-спектрометр построен А. Демпстером в 1918 г., а первый масс-спектрограф создал Ф. Астон в 1919 г.; он же исследовал изотопный состав большого числа элементов. Первый серийный масс-спектрометр создан А. Ниром в 1940 г.; его работы положили начало изотопной масс-спектрометрии. Прямое соединение массспектрометра с газо-жидкостным хроматографом (1959) дало возможность анализировать сложные смеси летучих соединений, а соединение с жидкостным хроматографом с помощью термораспылительного устройства (1983)− смеси труднолетучих соединений.
Любой масс-спектрометр сочетает комплекс процессов, протекающих в нем. Так, сначала происходит ионизация молекул с образованием газообраз-
Современные проблемы и методы биотехнологии. Учеб. пособие |
328 |
ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.3. Масс-спектрометрия в биотехнологии
ных ионов, затем идет разделение ионов, регистрация их масс и относительных количеств.
Каждый ион, который может быть положительным или отрицательным, характеризуется отношением его массы к заряду (m/z). В соответствии с этим отношением происходит разделение различных ионов. В масс-спектрометре исследуемое вещество переводится в газообразное состояние до или в процессе ионизации.
Macс-спектральные приборы. Для разделения ионов исследуемого вещества по величинам m/z, измерения этих величин и токов разделенных ионов используют масс-спектральные приборы.
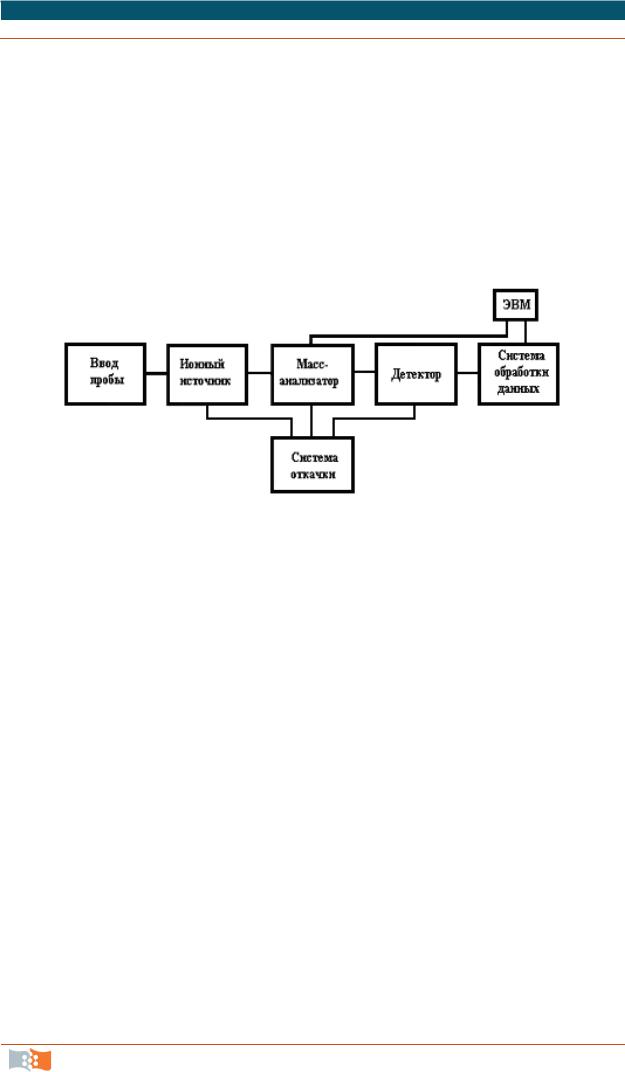
Рис. 6.15. Принципиальная схема масс-спектрометра
Масс-спектральные приборы состоят из системы ввода пробы (система напуска), ионного источника, разделительного устройства (масс-анализатора), детектора (приемника ионов), вакуумных насосов, обеспечивающих достаточно глубокий вакуум во всей вакуумной системе прибора, и системы управления и обработки данных (рис. 6.15). В современных приборах обработка данных и контроль задаваемых параметров производятся компьютером.
Превращения нейтрального атома или молекулы в заряженную частицу путем удаления из них или присоединения к ним одного или нескольких электронов называется ионизацией. Ионный источник предназначен для образования газообразных ионов исследуемого вещества и формирования ионного пучка, который направляется далее в масс-анализатор. Наиболее универсальный метод ионизации вещества − электронный удар. Впервые этот метод осуществлен П. Ленардом (1902). Современные источники такого типа построены по принципу источника А. Нира.
Для ионизации молекул обычно используют электроны с энергиями 70–100 эВ, которые движутся со скоростью 108 см/с и проходят путь, равный диаметру молекулы органического соединения за 10-16с. Этого времени до с- таточно для удаления электрона из молекулы вещества и образования молекулярного иона− положительно заряженного ион -радикала М+, имеющего энергию 2–8 эВ.
Современные проблемы и методы биотехнологии. Учеб. пособие |
329 |

ГЛАВА 6. СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ЦЕЛЕВЫХ ПРОДУКТОВ
6.3. Масс-спектрометрия в биотехнологии
Молекулярные ионы в зависимости от избытка внутренней энергии находятся в различных возбужденных состояниях, что обуславливает различное время их фрагментации или диссоциации. Обычно такой процесс протекает за время 10-13с. При этом молекулярные ионы превращаются в осколочные (фрагментные) ионы с более низкой электронной энергией. Фрагментные ионы, которые обладают еще достаточным избытком колебательной энергии, претерпевают дальнейший распад. Этот процесс продолжается до тех пора, пока не будет израсходован весь избыток внутренней энергии, присущий родоначальному молекулярному иону.
Образование фрагментных ионов характеризуется энергией (потенциалом) появления, т.е. минимальной энергией электронов, при которой начинает детектироваться соответствующий ион. Нетрудно представить, что энергия появления молекулярного иона есть не что иное, как энергия ионизации молекулы.
При распаде молекулярного иона, который является нечетноэлектронной частицей, образуются фрагментный ион и нейтральная частица, которая может быть как четно-электронной, так и нечетно-электронной, т.е. радикалом типа H•, •CH3 и т.д. При отщеплении нейтральной частицы возникает новый нечетно-электронный ион-радикал:
М+• → [Ф1]+• + N,
а при выбросе радикала − четно-электронный ион:
М+• → [Ф2]+ + N•.
Дальнейшие направления распада этих ионов различны. Нечетноэлектронный ион [Ф1]+• способен далее подобно М+• элиминировать либо нейтральную четно-электронную молекулу, либо радикал, в то время как катион [Ф2]+ в подавляющем большинстве случаев может терять лишь четноэлектронную молекулу (четно-электронное правило).
Таким образом, процессы фрагментации молекулярных ионов характеризуются значительным числом последовательных и конкурирующих направлений распада, вероятность которых определяется природой молекулы и элементов, ее составляющих, энергией разрывающихся связей, внутренней энергией образующихся ионов, стабильностью ионов и элиминирующихся нейтральных частиц, а также временным интервалом между образованием иона и его детектированием. Ионы с минимальным запасом энергии достаточно устойчивы и достигают приемника. Ионы с большим запасом внутренней энергии распадаются на пути движения на ионы с меньшей молекулярной массой (осколочные ионы), характерные для вещества определенного строения. Для ионизации молекул энергия электронного пучка должна превышать некоторую критическую для вещества величину, называемую потенциалом ионизации.
Современные проблемы и методы биотехнологии. Учеб. пособие |
330 |