2.2.3. Половые хромосомы
2.2.3.1. Первые наблюдения
Нерасхождение половых хромосом и определение пола у дрозофилы. В 1910 г. Морган [448] впервые детально описал Х-сцепленное наследование и X-Y-тип хромосомного определения пола у Drosophila melanogasler. В его опытах наблюдалось несколько исключений, не согласующихся с предположением о сцеплении с Х-хромосомой. Бриджес объяснил эти исключения, предположив возможность особых аномалий в механизме мейоза. В 1916 г. он показал [311], что в мейозе у дрозофилы действительно может иметь место нерасхождение половых хромосом. Дрозофила имеет четыре пары хромосом: три аутосомы и две половые хромосомы. Так же как и у человека, самки имеют набор XX, а самцы XY, т.е. все самцы по генам, сцепленным с Х-хромосомой, являются гемизиготными (полузиготными). Стало быть, каждая нормальная мужская гамета будет нести либо Х-, либо Y-хромосому, а все яйцеклетки Х-хромосому. В скрещиваниях самок, гомозиготных по Х-сцепленному признаку white (белоглазые), с самцами дикого типа (красноглазыми) все потомки-самцы, будучи гемизиготными, должны иметь белые глаза, как и их гомозиготные матери. Все дочери должны быть гетерозиготными и иметь нормальные красные глаза. Как правило, эти предположения оправдывались, однако в очень редких случаях самцы имели нормальные красные глаза, а самки белые. Бриджес показал, что это связано с нерасхождением материнских Х-хромосом, что приводит к образованию яйцеклеток либо с двумя Х-хромосомами, либо вообще без них (рис. 2.67). После оплодотворения спермиями самца дикого типа возможно образование зигот четырех типов: XXX; XXY; XO; YO. Зиготы YO не обнаружены, очевидно, в силу их нежизнеспособности. Остальные три типа зигот действительно обнаруживаются. Существование таких мух позволяет делать вывод относительно механизма определения пола:
![]() фенотип
самки
фенотип
самки
в) ХО фенотип самца (стерильный) Следовательно, фенотипический пол у плодовой мушки зависит от числа Х-хромосом. Одна Х-хромосома определяет пол самца, большее их число – пол самки. Y-хромосома также влияет на определение пола, поскольку самцы ХО стерильны.
Генотип ХО у мыши. Х-сцепленная мутация scurfy (sf) (шерсть животных как бы покрыта пылью) впервые была выявлена как спонтанная. Гемизиготные самцы стерильны, поэтому линию можно поддерживать только путем скрещивания гетерозигот (Xsf/X+) с нормальными самцами (X+/Y). Соотношение мутантных и нормальных самцов в таком скрещивании должно быть 1:1, а все самки в потомстве должны быть нормальными. Однако изредка появляются исключительные самки. Подобно самцам-гемизиготам, они стерильны. Яичники таких самок пересаживали нормальным самкам, которых затем скрещивали с самцами дикого типа. В потомстве от такого скрещивания все самцы были sf а все самки - нормальные. Фенотипически нормальные самки разделялись, однако, на две группы: одни из них передавали, а другие не передавали мутацию scurfy своим потомкам. Дальнейший анализ показал, что эти самки различаются генетически:
|
Рис. 2.67. Нерасхождение Х-хромосом у Drosophila, скрещивание самок white с самцами дикого типа. (Sinnott-Dunn-Dobzhansky, Principles of Genetics, 1958.) |
98 2. Хромосомы человека
Х+/О и X+/Xsf; самки с первым кариотипом не передавали sf в отличие от самок со вторым кариотипом. Этот эксперимент показывает, что особи ХО у мыши являются фертильными самками. Следовательно, у мыши за фенотипический пол ответственна не Х-хромосома, a Y-. Впоследствии ХО-кариотип у мыши обнаруживали довольно часто. В большинстве случаев это состояние возникает не вследствие мейотического нерасхождения, а в результате потери хромосомы после оплодотворения. При исследовании мутационного процесса утрата хромосомы стала важным инструментом для учета мутагенной активности (разд. 5.2). Позже у мыши был обнаружен и кариотип XXY. В отличие от дрозофилы, у которой особи XXY являются самками, у мыши особи XXY имеют фенотип самцов.
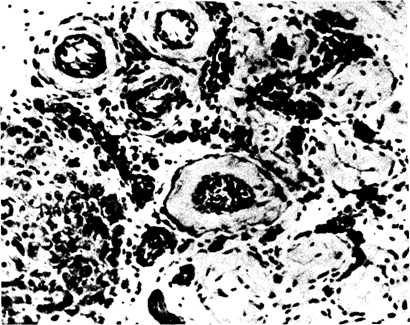
Анеуплоидия по Х-хромосоме у человека: XXY; XO; XXX. Анеуплоидия по Х-хромосоме была первой из выявленных у человека хромосомных аномалий. Когда Джекобс и Стронг (1959) [395] обследовали 42-летнего мужчину с типичными признаками синдрома Клайнфельтера (рис. 2.68; 2.69) (гинекомастия, маленькие тестикулы, гиалиноз тестикулярной ткани), они обнаружили Х-хроматин в клетках эпителия ротовой полости и «барабанные палочки» в нейтрофильных лейкоцитах. При исследовании хромосом в клетках костного мозга была выявлена добавочная субметацентрическая хромосома «в группе средних по размеру». Авторы предположили, что кариотип больного – XXY. Однако «не может быть исключена возможность... что добавочная хромосома представляет собой аутосому, несущую феминизирующие гены». Оба родителя больного имели нормальный кариотип с 46 хромосомами, следовательно, нерасхождение произошло у одного из них в мейозе. Вскоре после этого кариотип XXY был подтвержден при многих других случаях синдрома Клайнфельтера.
|
Рис. 2.68. Основные клинические симптомы синдрома Клайнфельтера. |

2. Хромосомы человека 99
|
Рис. 2.69. Гиалинизированная тестикулярная ткань при синдроме Клайнфельтера. Нормальные семенные трубочки отсутствуют и замещены гиалинизированной тканью. |
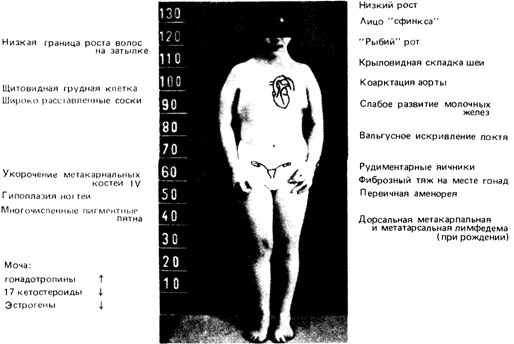
В то же самое время Форд и сотр. (1959) [352] выявили кариотип ХО. В этом случае 14-летняя девочка имела клинические признаки синдрома Тернера (рис. 2.70) при отсутствии в клетках эпителия слизистой оболочки рта Х-хроматина. Модальное число хромосом в клетках костного мозга было 45, обнаружено только 15 «метацентрических хромосом средней длины», как у нормальных мужчин. Это строго соответствовало кариотипу ХО. Сравнивая эти результаты с тем, что было известно для дрозофилы (рис. 2.67), авторы пришли к выводу, что в противоположность плодовой мушке тип ХО у человека приводит к развитию «агонадального» индивида с женским фенотипом. Упомянув о кариотипе XXX у дрозофилы, они отметили, что у человека он еще не описан.
Вскоре, однако, появилось сообщение о 35-летней женщине с плохо развитыми наружными половыми признаками и вторичной аменореей. Ее хромосомный набор состоял из 47 хромосом, причем добавочной была явно Х-хромосома: 47, XX, + X. В этом случае были исследованы и костный мозг, и фибробласты. Во многих клетках слизистой оболочки рта и в некоторых нейтрофилах удалось обнаружить два тельца Х-хроматина. Таким образом,
1) в противоположность дрозофиле фенотипический пол у человека определяется наличием или отсутствием Y-хромосомы, а не числом Х-хромосом. В этом отношении человек подобен мыши, но тип ХО у мыши соответствует фертильной самке, в то время как у человека – это женщина с нефункционирующими яичниками;
2) число телец Х-хроматина на одно меньше, чем число Х-хромосом.
Эти два факта стали фундаментом наших знаний и гипотез относительно детерминации пола и генетической активности Х-хромосом.
100 2. Хромосомы человека
|
Рис. 2.70. Основные клинические симптомы синдрома Тернера. |
2.2.3.2. Х-хромосо иные анеуплоидии у человека: современное состояние проблемы
Различие между Х-хромосомными и аутосомными анеуплоидиями. Вскоре после первых открытий анеуплоидии по половым хромосомам последовали и друше. Если рассматривать все эти случаи в целом как группу, то обнаруживаются заметные отличия от аутосомных анеуплоидии, о которых шла речь ранее:
1) умственное развитие в среднем хотя и ниже нормы, но аномалии развития мозга выражены не столь отчетливо, как при аномалиях аутосом. Многие пробанды имеют нормальное умственное развитие, а некоторые - даже выше среднего (разд. 8.2.2.2);
2) фенотипические нарушения в большей степени затрагивают развитие половых органов и гормонозависимый рост. Наблюдаются и другие пороки развития, особенно при синдроме Тернера, но встречаются они реже и по масштабам менее тяжелые.
В конечном счете Х-хромосомная анеуплоидия не нарушает эмбриональное развитие в такой степени, как аутосомные анеуплоидии. Объяснение, по-видимому, состоит в том, что в норме женщина имеет две, а мужчина только одну Х-хромосому, в связи с чем в эволюции сформировался специальный мощный механизм компенсации различий в дозе генов, сцепленных с Х-хромосомой. Этот же механизм оказался фактором, благоприятствующим носителям Х-хромосомных анеуплоидии.
Клиническая классификация Х-хромосомных анеуплоидии: мозаики. Наиболее важные численные и структурные аномалии Х-хромосом представлены в табл. 2.10. В общем, число добавочных Х-хромосом увеличивает степень умственной отсталости. Число телец Х-хроматина всегда на одно меньше,
2. Хромосомы человека 101
чем число Х-хромосом. В табл. 2.10 описаны также наиболее частые типы мозаицизма, однако обмены с участием Х-хромосомы не указаны. В последнем случае приложимы те же правила, что и для аутосомных транслокаций, в частности, в отдельных семьях наблюдается значительная вариабельность фенотипических проявлений.
Некоторые из Х-аутосомных обменов важны для разработки теоретических концепций относительно инактивации Х-хромосомы.
Крупные пороки развития обнаруживаются чаще всего при гонадальном дисгенезе, который является наиболее гетерогенным в клиническом и цитогенетическом отношении. Некоторые авторы, исходя из клинических принципов, предполагают возможность подразделения этого синдрома на варианты [88, 477]. Наиболее известная классификация основана на противопоставлении, с одной стороны, простого гонадального агенеза без добавочных симптомов, а с другой – синдрома Тернера с признаками, представленными на рис. 2.70. Однако онтогенетические данные слабо или вовсе не коррелируют с таким подразделением на две группы.
Анеуплоидия ХО встречается много реже, чем варианты XXY и XXX. Ожидаемые частоты различных типов зигот основаны на закономерностях распределения хромосом по гаметам после нерасхождения в первом или втором делениях мейоза у лиц обоего пола (рис. 2.71). При этом делается допущение, что вероятность оплодотворения любой яйцеклетки спермием X или Y составляет 1/2 и что все спермин (включая и анеуплоидные) участвуют в оплодотворении яйцеклеток. Эти события можно обозначить следующим образом:
NDF1 нерасхождение в первом мейотическом делении у женщин
NDF2 нерасхождение во втором мейотическом делении у женщин
NDM1 нерасхождение в первом мейотическом делении у мужчин
NDM2X нерасхождение, затрагивающее X-хромосому во втором мейотическом делении у мужчин
NDM2Y нерасхождение, затрагивающее Y-хромосому во втором мейотическом делении у мужчин
Таблица 2.10. Численные и структурные Х-хромосомные анеуплоидии у человека |
||
Кариотип |
Фенотип |
Приблизительная частота |
XXY |
Синдром Клайнфельтера |
1/700 мужчин |
XXXY |
Вариант синдрома Клайнфельтера |
~ 1/2500 мужчин |
XXXXY |
Глубокая умственная отсталость; сильное недоразвитие половых органов; радиоульнарный синостоз |
Очень редкий |
XXX |
Иногда легкая олигофрения, непостоянные нарушения функции гонад |
1/1000 женщин |
ХХХХ ХХХХХ |
Физически нормальные; тяжелая умственная отсталость |
Редкий |
Мозаики XXY/XY и XXY/XX |
Сходен с синдромом Клайнфельтера, иногда с более сглаженными симптомами |
~ 5-15% от больных с синдромом Клайнфельтера |
Мозаики ххх/хх |
Сходен с XXX |
Редкий |
ХО |
Синдром Тернера |
~ 1/2500 женщин при рождении |
Мозаики ХО/ХХ и ХО/ХХХ |
(Тернер); очень разные степени проявления |
Не редкий |
Различные структурные аномалии Х-хромосомы |
|
Не редки |
XYY |
Высокий рост; иногда аномалии поведения |
1/800 мужчин |
XXYY |
Высокий рост; в остальном сходен с синдромом Клайнфельтера |
Редкий |
102 2. Хромосомы человека
|
Рис. 2.71. Генотип половых клеток при нерасхождении Х- и Y-хромосом в первом и втором мейотическом делении. Подробности см. в тексте. |

Ожидаемые относительные частоты (вероятности, Р) различных типов зигот:
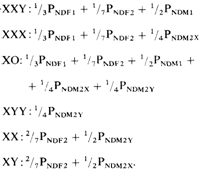
Следовательно, при отсутствии влияния других факторов и при условии равных вероятностей для NDM1 и NDM2X зиготы XXY должны встречаться несколько чаще, чем зиготы XXX. Существуют некоторые доказательства того, что нерасхождение аутосом происходит чаще в первом, чем во втором мейотическом делении (разд. 5.1.2.3).
Теоретически зиготы ХО должны встречаться чаще, чем зиготы других типов. Однако эмпирические данные не соответствуют этому, синдром Тернера встречается много реже, чем зиготы XXX и XXY. Это указывает на наличие сильного отбора против гамет, не содержащих половой хромосомы X, и/или на наличие сильного внутриутробного отбора против зигот ХО. Последнее предположение подтверждается тем, что среди спонтанно абортированных зародышей тип ХО встречается относительно часто. Есть и другие доказательства: риск нерасхождения обычно увеличивается с возрастом матери (разд. 5.1.2.2). Для анеуплоидий типа XXX и XXY в отличие от типа ХО эта закономерность выявляется вполне четко. В настоящее время допускается, что выжившие зиготы ХО являются результатом не мейотического, а ми готического нерасхождения или утраты хромосом на ранних стадиях дробления. Об этом же свидетельствуют данные, что в группе больных с синдромом Тернера мозаики встречаются относительно чаще, чем в группах XXX- и XXY-анеусомиков. С другой стороны, зиготы XYY могут возникать только вследствие нерасхождения во втором мейотическом делении у мужчин. Однако они встречаются почти так же часто, как и зиготы XX Y. Следовательно, вероятность нерасхождения по Y-хромосоме, PNDM2Y, явно много выше, чем отдельные вероятности разных типов нерасхождения по Х-хромосоме. Наблюдались мозаики всех типов. Механизмы возникновения мозаиков обсуждаются в разд. 5.1.6.
Интерсексы. На основании клинических данных различают три типа интерсексов: 1) истинный гермафродитизм: присутст-
2. Хромосомы человека 103
вуют половые клетки обоих полов;
2) мужской псевдогермафродитизм: имеются только тестикулы;
3) женский псевдогермафродитизм: имеются только яичники.
К сожалению, эта простая классификация не совпадает с цитогенетическими данными, поскольку в каждой группе встречаются хромосомные варианты, включая и такие случаи, как, например, 46,ХХ у мужчин. Многие интерсексы являются мозаиками, содержащими клетки с различным набором половых хромосом в разных комбинациях. Например, фенотип мозаиков 45,XX/46,XY может проявиться в виде овариального дисгенеза, гонадального дисгенеза, с мужским псевдогермафродитизмом или в форме «смешанного гонадального дисгенеза», когда одна гонада представлена фиброзным тяжем, а другая – диспластическим тестикулом. Некоторые истинные гермафродиты имеют кариотип 46,XX/46,XY. Мозаицизм типа XX/XY может возникать как следствие любого из девяти различных механизмов, таких, как оплодотворение ооцита двумя различными спермиями, слияние двух оплодотворенных яйцеклеток, митотическая ошибка во время первого дробления или внутриутробный обмен стволовыми кроветворными клетками между разнополыми дизиготными близнецами (разд. 3.8.3).
Первичная функция полоопределяющих факторов состоит в индукции гонад. Гонады в свою очередь детерминируют развитие других половых органов и вторичные половые признаки. Нарушения индукции гонад могут быть вызваны либо аномалиями набора половых хромосом, либо другими факторами, не имеющими прямого отношения к половым хромосомам. В последнем случае интерсексы могут иметь нормальный набор хромосом как XX, так и XY. Сбалансированные структурные перестройки с участием Х-хромосомы часто ведут к бесплодию у обоих полов [427].
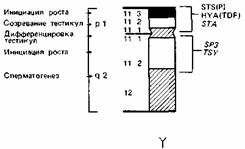
Y-хромосома как фактор, определяющий мужской пол. Развитие тестикул детерминируется Y-хромосомой. Бюлер [315], обобщив данные многочисленных литературных сообщений о больных с аномалиями половой дифференцировки и структурными аномалиями Y-хромосомы, попытался локализовать различные функции в эухроматическом районе Y-хромосомы человека (рис. 2.72). На этом рисунке отсутствует, однако, информация, полученная на основе использования специфических ДНК-зондов. Различные типы аномалий Xи Y-хромосом весьма информативны относительно механизмов полового развития человека. Однако полезные сведения по этой проблеме можно получить также и при изучении метаболических аномалий с простым типом наследования, из опытов по получению химер у животных, а также из исследований HY-антигена. Кроме того, для полного понимания процесса половой дифференцировки необходимы знания о генетическом контроле регуляторных функций гормонов. В связи с этим особое значение приобретает концепция рецепторных болезней. Обсуждение этих вопросов будет представлено в разд. 4.7.5, здесь же мы обсудим один специальный вопрос – дозовую компенсацию.
|
Рис. 2.72. Слева: функции, аномалии которых отмечены у больных с аберрациями Y-хромосомы [315]. Справа: локализация Y-специфических зондов ДНК (из каталога McKusick 1985). |