

2.1.2. Кислотно-основное титрование
Решение вопроса о соответствии характерной точки на кривой потенциометрического титрования, принимаемой за КТТ (точка перегиба на интегральной кривой или точка максимума на дифференциальной кривой), моменту эквивалентности всегда необходимо провести анализ уравнения кривой титрования. Вывод такого уравнения и его вид зависят от типа изучаемой реакции. Рассмотрим подробно такую задачу на примере протолитических реакций.
Кривые потенциометрического кислотно-основного титрования E = f(y) (где y - концентрация добавленного титранта) являются отображением изменения в ходе титрования рН раствора. Поэтому в качестве индикаторного электрода может быть выбран любой из перечисленных выше рН-чувствительных электродов, потенциал которого зависит от концентрации ионов водорода. Чаще всего используют стеклянный электрод.
Для упрощения теоретического расчета кривых титрования обычно оперируют концентрациями, а не активностями ионов, т.е. допускают, что растворы разбавлены и коэффициенты активности компонентов можно принять равными 1, или в растворе присутствует индеферентный электролит, который обеспечивает постоянство коэффициентов активности в ходе титрования. В этом случае можно оперировать величиной pH= −lg[H+], уравнение потенциала индикаторного электрода можно записать в виде:
![]() (2.1)
(2.1)
Для всех водородных электродов эти уравнения будут отличаться только величиной стандартного потенциала Eo, поэтому полученные с помощью разных электродов кривые потенциометрического титрования будут идти симбатно.
Кроме того при расчетах обычно пренебрегают разбавлением раствора в ходе титрования. При этих допущениях экспериментальные кривые не совпадают с теоретически рассчитанными кривыми. Однако возникающая при этом небольшая погрешность не имеет существенного значения при определении рН конечной точки титрования.
Форма кривой и положение точки эквивалентности зависят от силы кислоты или основания. Для любой точки потенциометрической кривой потенциал электрода можно рассчитать по формулам для вычисления рН растворов соответствующих кислот и оснований. В случае кислот (оснований), которые полностью диссоциируют в растворе, необходимые данные можно получить сравнительно просто. Например, при титровании сильной кислоты сильным основанием, которое протекает согласно схеме
HA + BOH BA + H2O, (2.2)
кривую титрования pH=f(y) легко получить на основании принципа электронейтральности растворов, который для представленной схемы реакции можно представить в виде равенства
[H+] + [B+] = [A-] + [OH-]. (2.3)
Учитывая выражение ионного произведения воды Kw=[H+][OH-] и численное равенство концентраций введенного основания и катионов металла при титровании кислоты, запишем
y = [B+] = [A-] − [H+] + Kw / [H+]. (2.4)
В ходе титрования сильной кислоты сильным основанием концентрация анионов будет оставаться постоянной (если пренебречь разбавлением раствора) и равной исходной концентрации кислоты Co. Решив квадратное уравнение
y = Co − [H+] + Kw / [H+] , (2.5)
получим выражение для расчета концентрации ионов водорода в ходе титрования
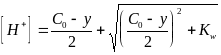 .
(2.6)
.
(2.6)
По найденным значениям [H+] можно рассчитать величины pH и потенциал индикаторного электрода E при заданных значениях y, т.е. рассчитать кривую титрования. Для момента эквивалентности, когда y=C0
![]() ,
а pHy=Co = 7. (2.7)
,
а pHy=Co = 7. (2.7)
Чтобы оценить форму кривой нейтрализации и выбрать на ней характерную точку, отвечающую конечной точке титрования, необходимо проанализировать уравнение (2.5), т.е. найти выражения первой производной dpH/dy (так называемая “податливость” системы), второй производной d2pH/dy2 и условия их равенства нулю.
На практике чаще пользуются величиной обратной “податливости”, которую называют “буферной емкостью” (или для краткости “буферностью”)
Р = dy/dpH = d[B+]/dpH . (2.8)
Дифференциировать уравнение (2.5) по pH для нахождения буферности нельзя. Учитывая, что
dpH = d(-lg[H+]) = -0,43(dln[H+]) = - d[H+] / 2,3[H+]. (2.9)
выражение для “буферности” можно получить, умножая уравнение (2.5) на −2,3[H+] и дифференциируя по [H+]:
![]() .
(2.10)
.
(2.10)
Из последнего соотношения следует, что буферность растворов сильных протолитов ни при каких значениях pH не может равняться нулю, поэтому кривая нейтрализации не имеет экстремума.
Чтобы выяснить имеет ли кривая титрования точку перегиба, необходимо выяснить как ведет себя буферность с изменением pH, т.е. найти выражение производной dP/dpH. Дифференциируя уравнение (2.10) с учетом (2.9), получим
![]() .
(2.11)
.
(2.11)
Эта производная равна нулю, если (1- Kw/[H+]2)=0 или [H+]=(Kw)1/2=10-7 моль/л. При этом условии кривая P = f(pH) проходит через экстремум – минимум; что это действительно минимум можно убедиться еще раз продифференцировав уравнение (2.11) и установив положительное значение второй производной. Кривая титрования pH=f(y) через точку перегиба, координаты которой отвечают условиям эквивалентности.
Задавая различные значения [H+] или pH растворов, содержащих только сильные электролиты, можно по уравнению (2.10) рассчитать значения буферности и построить диаграмму буферности P=f(pH). Результаты расчетов представлены в табл. 2 и на рис. 2.1.
Таблица 2. Расчет буферности водных
растворов сильных протолитов.
[H+] pH P=2,3([H+]+[OH-])
1 0 2,30 110-1 1 2,30.10-1 1.10-2 2 2,30.10-2 4.10-3 2,4 1,00.10-2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.10-6 6 2,32.10-6 1.10-7 7 4 60.10-7 1.10-8 8 2,32.10-6 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2,5.10-12 11,6 1,00.10-2 1.10-12 12 2,30.10-2 1.10-13 13 2,30.10-1 1.10-14 14 2,30 |
|
|
Рис. 2.1. Диаграмма буферности водных растворов сильных протолитов. |
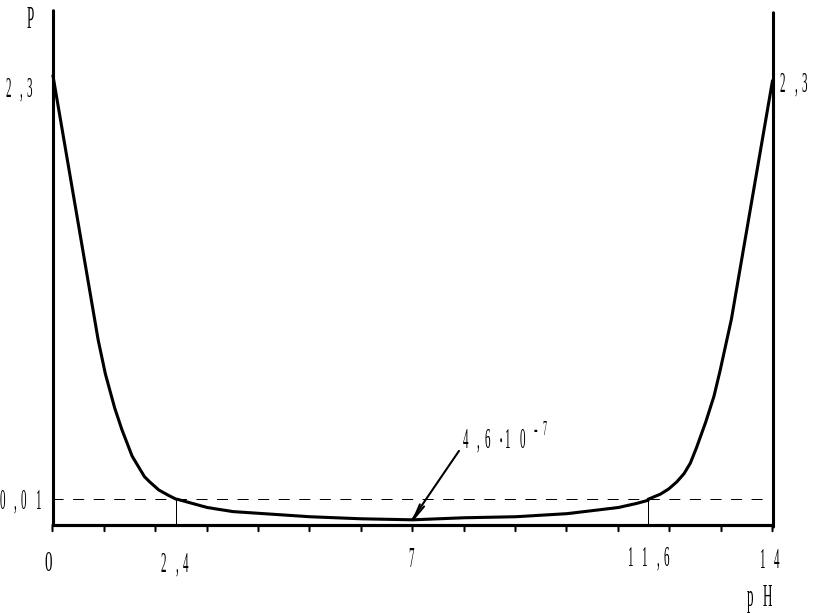
Как видно, при добавлении щелочи к раствору сильной кислоты буферность сначала уменьшается, достигая минимального значения 4,6.10-7 при pH=7, затем снова возрастает, причем в широком интервале pH от 2,4 до 11,6 буферность имеет весьма низкое значение (менее 0,01).
Согласно диаграмме бу-ферности, кривая титрования pH=f(y) S-образной формы с быстрым изменением рН (скачком рН) в области 2,4 – 11,6 единиц и точкой перегиба с координатами pH=7 и y=C0 (рис.2.2). При уменьшении исходной концентрации анали-зируемой кислоты скачок становится более размытым, поэтому минимальные опреде-ляемые концентрации порядка |
|
10-4 моль/л. |
Рис.2.2. Кривые титрования сильной кислоты различной концентрации сильным основанием |
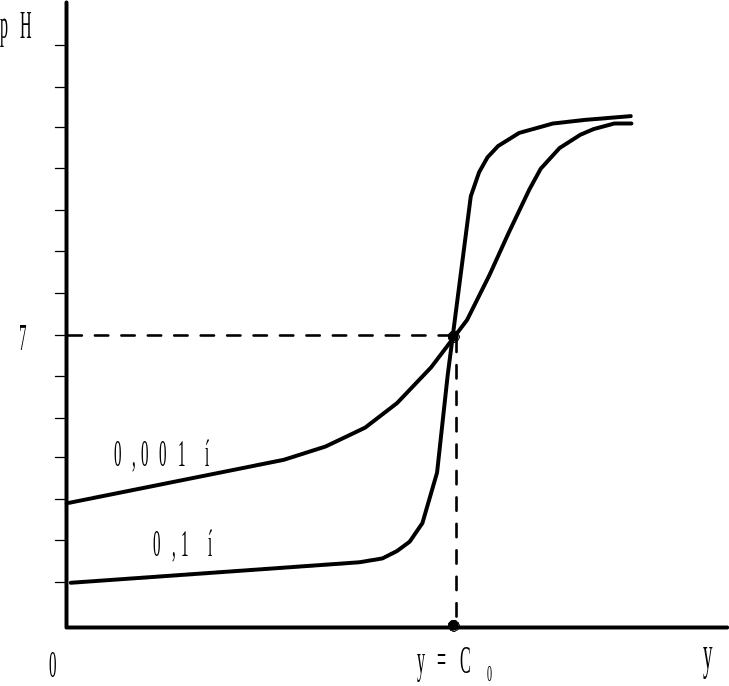
Нейтрализации смесей сильных кислот, полностью диссоциирующих в водной среде, отвечает та же диаграмма буферности и та же форма кривой титрования с одним скачком рН, регистрируя который можно определить лишь суммарное содержание кислот в растворе.
Нейтрализация слабой кислоты сильным основанием характеризуется тем, что концентрация анионов в ходе титрования не остается постоянной. Если представить уравнение константы диссоциации слабой кислоты в виде
![]() (2.12)
(2.12)
и учесть, что концентрация непродиссоциировавшей кислоты [HA]=C0-[A-], то можно записать
![]() .
(2.13)
.
(2.13)
Подставляя полученное выражение в уравнение (2.4), получим уравнение кривой титрования слабой кислоты сильным основанием
![]() .
(2.14)
.
(2.14)
Это уравнение третьей степени и точный расчет [H+] затруднен.
На практике поступают обычно проще. В начальный момент титрования, когда y = 0, справедливо равенство
![]() .
(2.15)
.
(2.15)
Величина KW/[H+] на несколько порядков меньше первого слагаемого правой части уравнения (2.15), поэтому ей можно пренебречь. Тогда
![]() .
(2.16)
.
(2.16)
Решая это квадратное уравнение, имеем
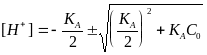 .
(2.17)
.
(2.17)
Если С0КА, то хорошим приближением концентрацию ионов водорода в исследуемом растворе кислоты до начала титрования можно рассчитать по формуле
![]() .
(2.18)
.
(2.18)
Для недотитрованных растворов, когда y C0, концентрацию ионов водорода рассчитывают, используя уравнение константы диссоциации
![]() .
(2.19)
.
(2.19)
Пренебрегая диссоциаций слабой кислоты в присутствии ее соли, можно считать, что равновесная концентрация [HA][недотитрованной кислоты], а равновесная концентрация анионов [A-] [образовавшейся соли]. Тогда
![]() (2.20)
(2.20)
или
![]() .
(2.21)
.
(2.21)
В точке эквивалентности, когда y=C0, вследствие гидролиза образовавшейся соли
В+А- + H2O = HA + B+OH-
раствор будет не нейтральным, а щелочным.
Концентрация ионов водорода может быть
вычислена по известному уравнению
![]()
![]() ,
(2.22)
,
(2.22)
а рН раствора по уравнению
![]() .
(2.23)
.
(2.23)
Концентрацию соли в эквивалентной точке С можно приравнять исходной концентрации кислоты С0, если не учитывать разбавление раствора в ходе титрования.
Для того чтобы выяснить форму кривой титрования слабой кислоты и имеющиеся характерные точки на этой кривой, проанализируем уравнение (2.14), воспользовавшись опять величиной буферности
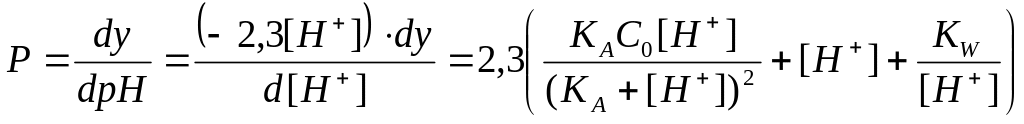 .
(2.24)
.
(2.24)
Эта производная, ни при каких значениях рН не может равняться нулю, следовательно, кривая титрования не должна иметь экстремум. Не проводя расчетов буферности, выясним как будет вести себя зависимость P=f(pH). Для этого необходимо продифференцировать уравнение (2.24) по рН. Учитывая, что в области недотитрованных (yC0) растворов слабой кислоты (КА≤10-5) выполняется неравенство
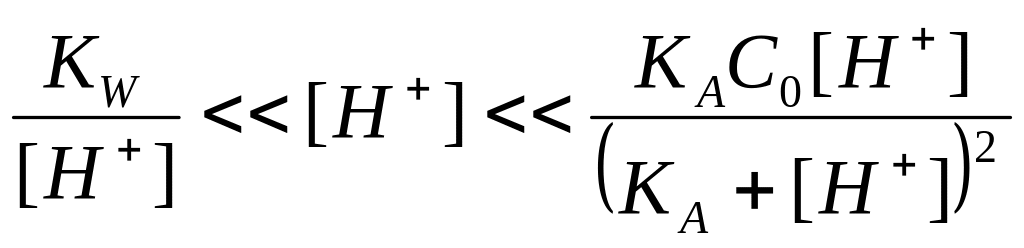 ,
(2.25)
,
(2.25)
уравнение (2.24) можно записать в виде
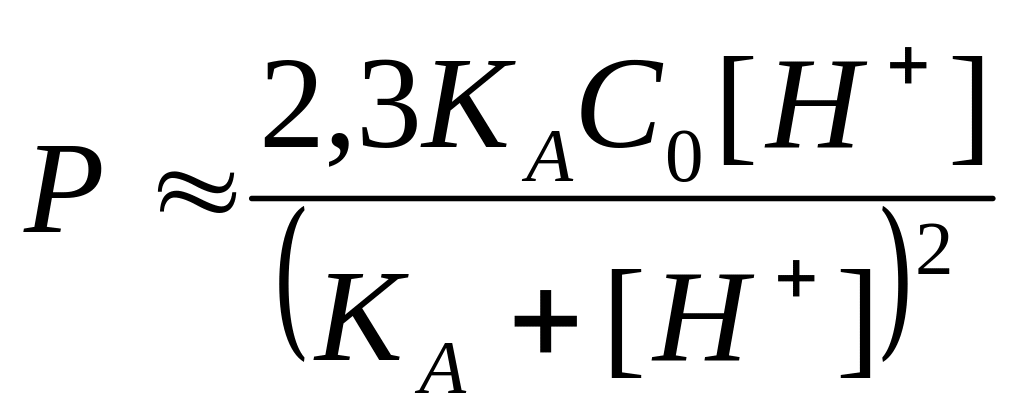 .
(2.26)
.
(2.26)
Дифференцируя последнее выражение по рН, получим
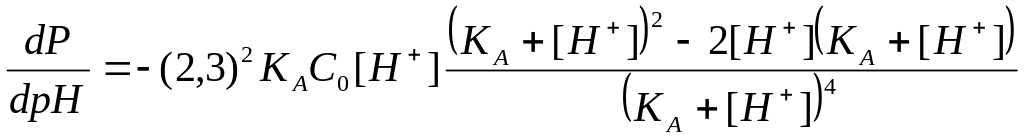 . (2.27)
. (2.27)
Производная dP/dpH=0, если [H+]=KA или pH=pKA. Этому значению pH на кривой P=f(pH) отвечает максимальное значение буферности Pmax, которое согласно уравнению (2.26) имеет значение
![]() (2.28)
(2.28)
и не зависит от силы кислоты, а определяется только ее концентрацией.
Равенство [H+]=KA, в соответствии с уравнением (2.20), отвечает условию y=0,5C0, т.е. полуоттитрованному раствору слабой кислоты. Если, например, сильным основанием титруется 0,1 н. слабая кислота, у которой КА=10-5, то изменение буферности системы в ходе титрования можно представить в виде кривой abdefg, приведенной на рис. 2.3. В данном случае наблюдается два участка повышенной буферности, первый из которых (bde) соответствует области недотитрованного раствора и обусловлен присутствием слабой кислоты и ее соли, а второй участок (fg) − избытком титранта.
|
|
Рис. 2.3. Изменение буферности при титровании слабой кислоты сильным основанием |
Рис. 2.4. Кривая титрования слабой кислоты (КА=10-5)сильным основанием |
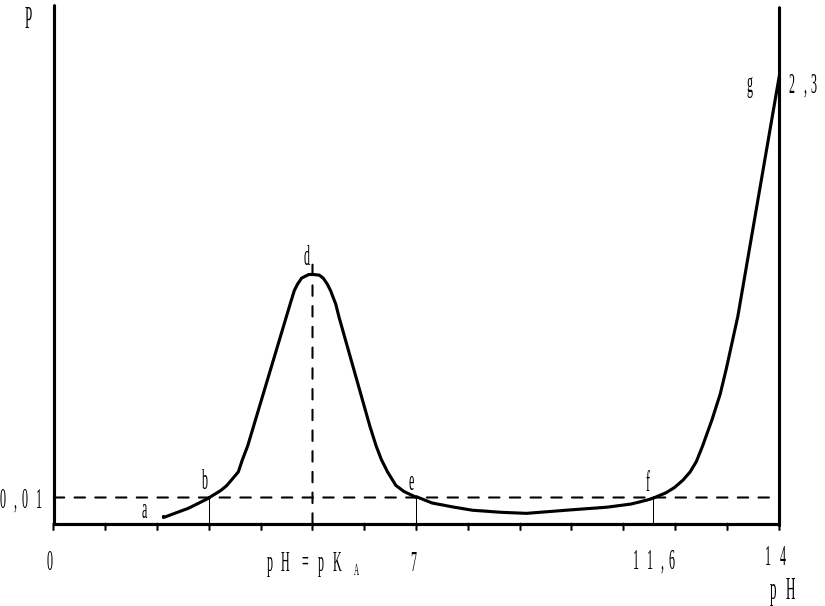

Согласно диаграмме буферности на кривой титрования pH=f(y) слабой кислоты, представленной на рис. 2.4, полуоттитрованному раствору, отвечает точка перегиба, ордината которой соответствует рН=рКА. Последнее равенство позволяет достаточно просто определять методом потенциометрического титрования концентрационные константы слабых кислот.
Протяженность участков низкой буферности, наблюдающихся в начале (участок ab) и в конце титрования (участок ef), зависит от силы кислоты. Чем слабее кислота (чем меньше КА), тем больше изменения рН в начале титрования и тем меньше протяженность участка низкой буферности (ef), который обуславливает величину скачка на кривой титрования.
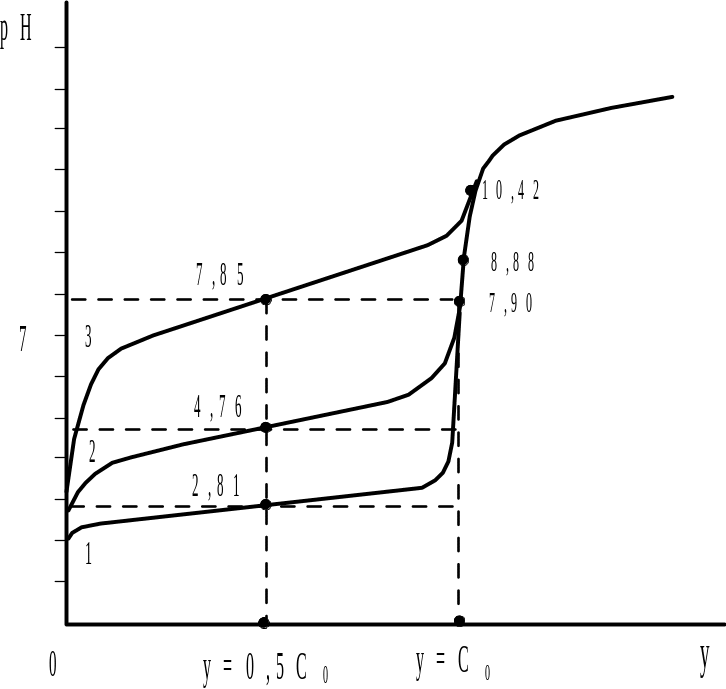
На рис. 2.5 представлены кривые титрования трех слабых кислот: хлоруксусной, уксусной и дихлорфенола, рКА которых равны соответственно 2,81, 4,76 и 7,85. Как видно, с уменьшением силы указанных кислот величина скачка рН уменьшается, а рассчитанное с учетом гидролиза рН в точке эквивалентности увеличивается. Если константы диссоциа-ции кислот малы, то в точке эквивалентности вообще не наблюдается скачка потенциала или перегиба на кривой титрования. Возникает вопрос − |
|
− какое минимальное значение константы диссоциации может иметь кислота, чтобы ее содержание можно было определить титрованием в водной среде? |
Рис. 2.5. Кривые титрования слабых кислот: 1)−хлоруксусная (рКА=2,81); 2)−уксусная (рКА=4,76); 3−2,4 дихлорфенол (рКА=7,85) |
Допустим, мы титруем слабую кислоту и добавили такую порцию титранта, когда нейтрализованной оказалась 0,1% первоначального количества кислоты. Тогда, согласно уравнению (2.21), рН анализируемого раствора имеет значение
![]() .
.
Титрование можно считать законченным, если оттитровано 99,9 % (максимальная точность объемного анализа) исходной кислоты. Тогда рН анализируемого раствора
![]() .
.
Таким образом, при определении слабой кислоты с точностью 0,1%, мы должны учитывать, что изменение рН раствора в ходе титрования составляет 6 единиц
![]() .
(2.29)
.
(2.29)
Если титруется 0,1 н. кислота с рКА=8,6, то она практически полностью будет нейтрализована при рН=11,6. Рассчитав для данного рН по уравнению (2.24) буферность системы, образованной слабой кислотой и сильным основанием, можно убедиться, что значение буферности практически будет равно 0,01. В этом случае участок (ef, рис. 2.3) с пониженной буферностью отсутствует, поэтому скачок рН в области точки эквивалентности отсутствует и на кривой титрования можно зарегистрировать только точку перегиба. Таким образом, количественное определение слабой кислоты в водных растворах с точностью 0,1% возможно, если выполняется условие
![]() .
(2.30)
.
(2.30)
Более слабые кислоты в водной среде титрованием определить невозможно.
Т.к. в уравнение (2.24) входит концентрация кислоты С0, то более точно условие определимости можно записать в виде
![]() .
(2.31)
.
(2.31)
Если удовлетворяет точность анализа 1%, то условие возможности определения слабой кислоты в водных растворах можно записать следующим образом
![]() (2.32)
(2.32)
или с учетом исходной концентрации кислоты
![]() .
(2.33)
.
(2.33)
Титрование смесей протолитов наиболее удобно проводить потенциометрическим методом, поскольку он позволяет обеспечить их раздельное определение в водной и неводной средах.
Если раствор в сравнимых концентрациях содержит несколько кислот различной силы, то при алкалиметрическом титровании эти кислоты взаимодействуют с сильным основанием в порядке увеличения рКА. Рассмотрим случай, когда в воде титруют смесь двух одноосновных кислот HA(1) HA(2) различной силы, причем рКА,1< рКА,2, а начальная концентрация этих кислот составляет соответственно С0,1 и С0,2. Исходя из принципа электронейтральности раствора, будет справедливо равенство
y = [B+] = [A(1)-]+[A(2)-] − [H+] + Kw / [H+], (2.34)
а учитывая диссоциацию каждой из кислот, уравнение кривой титрования можно записать в виде
![]() . (2.35)
. (2.35)
Не проводя расчета кривой титрования [H+]=f(y), ограничимся рассмотрением только ее формы и имеющихся на ней характерных точек. Для этого продифференцируем уравнение (2.34) по рН и получим выражение буферности
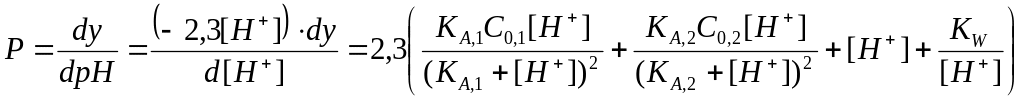 ,
(2.36)
,
(2.36)
из которого следует, что буферность свойство аддитивное и определяется присутствием всех протолитов в растворе.
Допустим, что константа диссоциации в воде более сильной кислоты КА,1=10-4,5 (рКА,1=4,5), а более слабой − КА,2=10-8,5 (рКА,2=8,5). Если задаваться различными значениями [H+], то можно рассчитать изменение буферности раствора смеси кислот в ходе титрования сильным основанием, которое описывается кривой, представленной на рис. 2.6. На данной диаграмме имеется две области с повышенной буферностью, которые обусловлены присутствием в анализируемом растворе двух буферных систем, образованных слабыми кислотами и их солями. При рН>11,6 высокая буферность связана с избытком титранта (сильного основания).
|
|
Рис. 2.6. Изменение буферности в ходе титрования водного раствора двух слабых кислот сильным основанием |
Рис. 2.7. Кривая титрования смеси двух слабых кислот сильным основанием в воде |
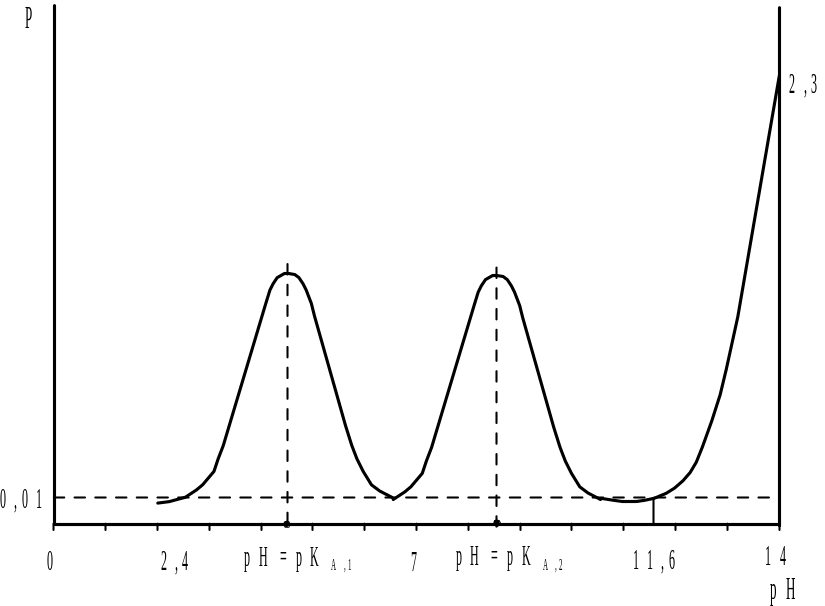
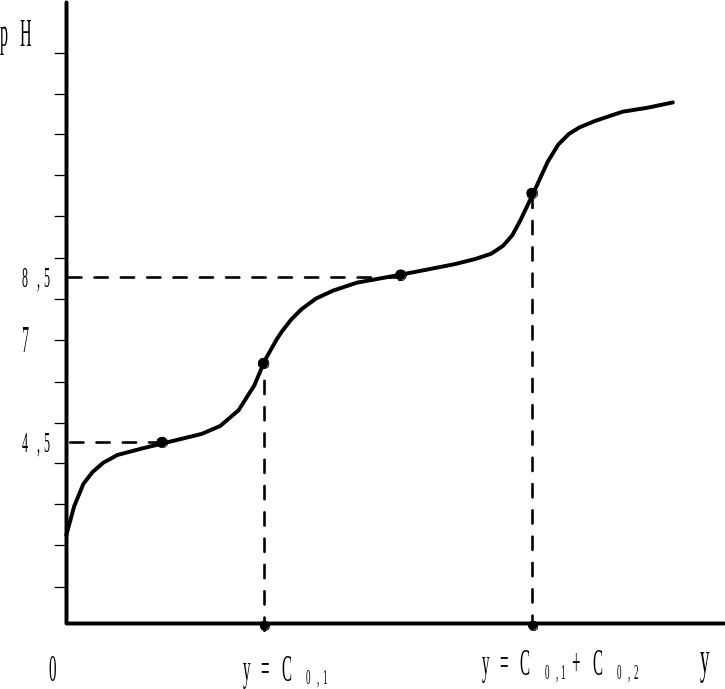
В соответствии с изменением буферности данной системы кривая титрования (рис. 2.7) имеет четыре точки перегиба, две из которых наблюдаются при значениях рН=рКА,1=4,5 и рН=рКА,2=8,5 и отвечают полуоттитрованному состоянию каждой из определяемых кислот. Две другие точки перегиба на кривой титрования соответствуют минимальным значениям буферности системы и служат для определения конца титрования.
На практике при анализе смеси слабых протолитов важно знать ответ на три вопроса:
как должны различаться константы ионизации, чтобы дифференцировано определить кислоты (основания);
какова точность анализа;
как рассчитать рН раствора в точках эквивалентности.
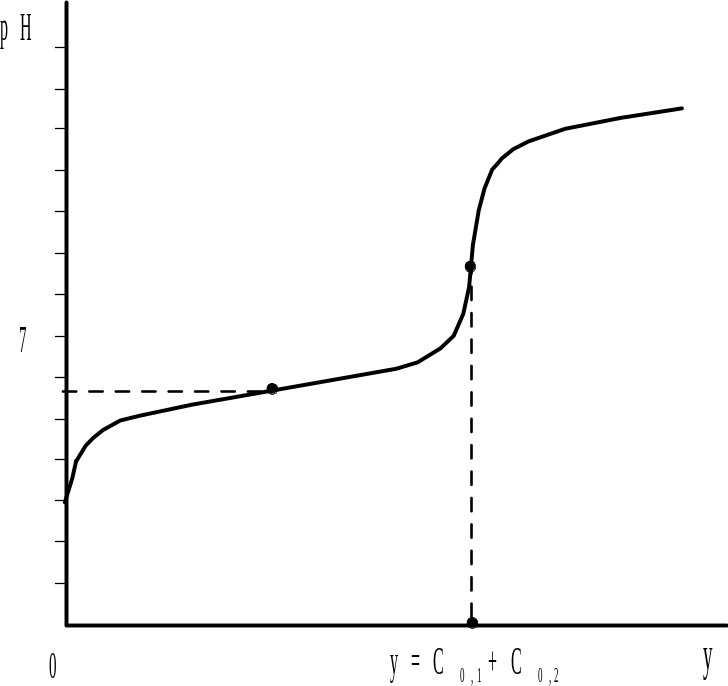
Титрование смеси кислот (оснований) приблизительно равной силы (рис. 2.8) не позволяет дифференцировано определить содержание каждого протолита из-за того, что области повышенной буферности, обусловленные присутствием этих протолитов, перекрываются, а концентрация ионов водорода в каждый момент титрования соответствует суммарному содержанию всех кислот и оснований в растворе. В этом случае при определенных условиях (для наиболее слабой кислоты должны выполняться уравнения (2.30) и (2.31) на кривой титрования (рис. 2.9) наблюдается только один скачок, по которому можно определить только суммарное содержание протолитов.
|
|
Рис. 8. Изменение буферности в ходе титрования смеси двух кислот с близкими константами диссоциации |
Рис. 9. Кривая титрования смеси двух кислот с близкими константами диссоциации |
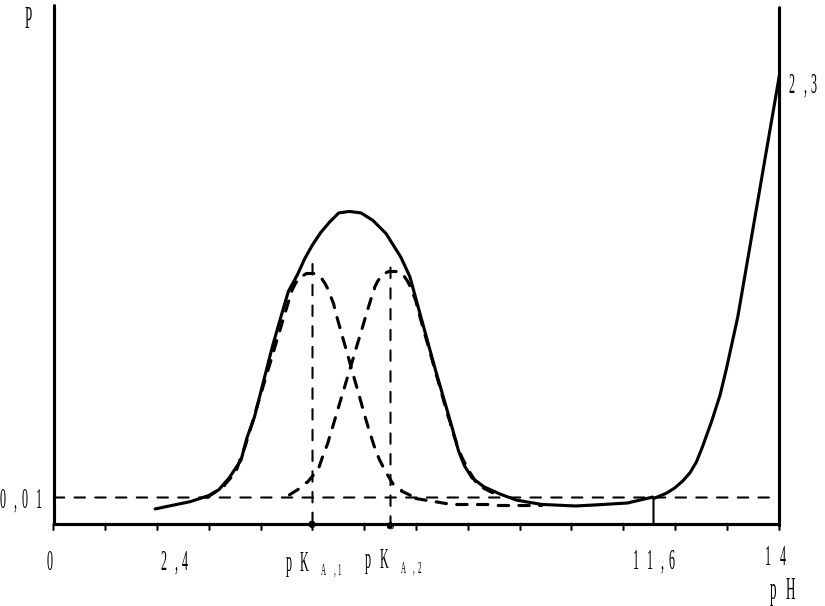
Анализ смеси кислот (оснований) в водной среде возможен лишь в тех случаях, когда соответствующие константы диссоциации титруемых компонентов, как это показано на рис. 2.6, отличаются друг от друга по меньшей мере на четыре порядка:
КА,1 / КА,2 ≥104 или ΔрК= рКА,2 -рКА,1 ≥4. (2.37)
В этих условиях обеспечивается достаточно заметное изменение потенциала индикаторного электрода вблизи первой точки эквивалентности и погрешность не превышает 1%, т.к. при рН= рКА,1+2 будет оттитровано 99% более сильной кислоты и только 1% более слабой кислоты.
Проведение определений, при которых погрешность не превышает ±0,1% , возможно при соблюдении более жестких условий:
КА,1 / КА,2≥106 или ΔрК= рКА,2 -рКА,1 ≥ 6. (2.38)
Если концентрации анализируемых кислот существенно различаются, то условия их дифференцированного определения необходимо записать в виде
С0,1КА,1 : С0,2КА,2 ≥106, (2.39)
при этом для наиболее слабой должно выполняться уравнение (2.31):
С0,2КА,2 ≥10-8,6.
Для того чтобы у точки эквивалентности появился скачок (или надежно регистрируемая точка перегиба) требуются 6 единиц шкалы рН. При наличии на кривой титрования 2 скачков у двух точек стехиометричности требуется 12 единиц рН. Однако нормальная шкала рН водных растворов имеет всего только 14 единиц рН. Следовательно, не превышая погрешности ±0,1%, в водных растворах последовательно можно титровать не более двух протолитов.
Если известны константы ионизации кислот, можно легко рассчитать ошибку их определения и рН для обеих точек стехиометричности. Когда в растворе присутствует две кислоты, то уравнение (2.12) справедливо для каждой из них
![]() и
и
![]() .
(2.40)
.
(2.40)
Концентрация иона водорода в обоих уравнениях одна и та же величина, поэтому можно написать равенство
![]() .
(2.41)
.
(2.41)
Другим словами, в любой момент титрования смеси двух кислот выполняется уравнение
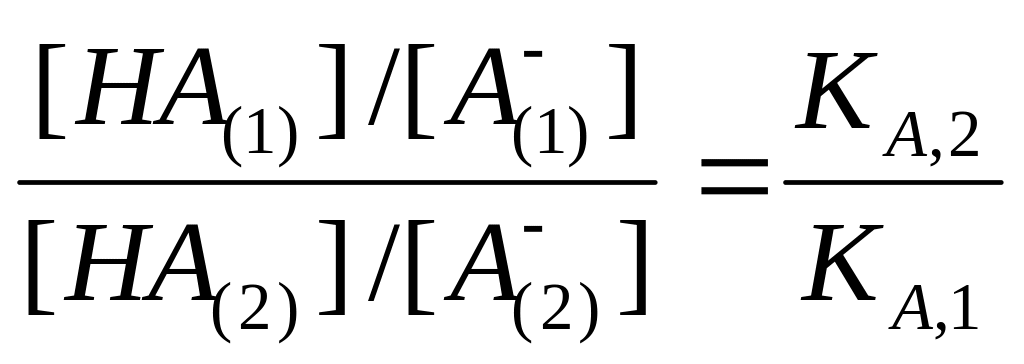 .
(2.42)
.
(2.42)
Если пренебречь диссоциацией слабой кислоты в присутствии ее соли, то равновесную концентрацию молекулярной формы кислоты можно считать концентрацией не нейтрализованной кислоты, а равновесную концентрацию аниона − концентрацией нейтрализованной кислоты.
Допустим, что КА,1 ≥
КА,2, С0,1=1,
![]() и к исходной смеси добавлено количество
щелочи эквивалентное содержанию более
сильной кислоты. В этот момент, поскольку
отношение КА,1 / КА,2
конечная величина, некоторое количество
более сильной кислоты z
будет не нейтрализовано и соответствующее
количество щелочи пойдет на нейтрализацию
более слабой кислоты. Величина z
будет определять ошибку анализа,
используя которую, уравнение (2.41) можно
записать в виде
и к исходной смеси добавлено количество
щелочи эквивалентное содержанию более
сильной кислоты. В этот момент, поскольку
отношение КА,1 / КА,2
конечная величина, некоторое количество
более сильной кислоты z
будет не нейтрализовано и соответствующее
количество щелочи пойдет на нейтрализацию
более слабой кислоты. Величина z
будет определять ошибку анализа,
используя которую, уравнение (2.41) можно
записать в виде
![]() .
(2.43)
.
(2.43)
Т.к. на практике интересуют случаи, когда
![]() ,
то для расчета абсолютной ошибки
используют уравнение
,
то для расчета абсолютной ошибки
используют уравнение
![]() ,
(2.44)
,
(2.44)
из которого следует, что ошибку анализа определяют не только константы ионизации, но концентрации протолитов. Причем, чем больше концентрация более слабой кислоты, тем больше ошибка.
Расчет рН характерных точек кривой титрования смеси слабых кислот (рис. 2.7) проводят следующим образом. В начальной точке (y=0) рН титруемого раствора определяется протолизом наиболее сильной кислоты HA(1) согласно уравнению (2.18)
![]() .
(2.45)
.
(2.45)
Когда оттитрована половина более сильной кислоты (y=1/2C0,1) рН=рКА,1. Для первой точке стехиометричности (y=C0,1) рН раствора можно рассчитать из уравнения (2.41), которое с учетом абсолютной ошибки определения z можно переписать в виде
![]()
или![]()
![]() .
(2.46)
.
(2.46)
Логарифмируя, получим
![]() .
(2.47)
.
(2.47)
Если С0,1=С0,2 , то рН в первой точке эквивалентности равно полусумме рК определяемых кислот
![]() .
(2.48)
.
(2.48)
Когда оттитровано половина более слабой кислоты (y=C0,1+0,5C0,2) рН = рКА,2. Водородный показатель для второй точки эквивалентности рассчитывают по уравнению (2.22), используя константу диссоциации более слабой кислоты
![]() .
(2.49)
.
(2.49)
При определении концентраций сильной и слабой кислоты в их смеси точность анализа зависит от константы диссоциации последней. С точностью ±0,1 % концентрацию каждой из кислот можно установить лишь в том случае, когда для слабой кислоты выполняется условие
10-5,4 ≥ С0,2КА,2 ≥10-8,6 или 5,4 ≤ рКА,2-lgC0,2 ≤ 8,6. (2.50)
Анализ с погрешностью ±1 % может быть выполнен при условии
10-4,4 ≥ С0,2КА,2 ≥10-9,6 или 4,4 ≤ рКА,2-lgC0,2 ≤ 9,6. (2.51)
Изменение буферности в ходе титрования смеси сильной и слабой кислот и кривая титрования представлены, соответственно, на рис. 2.10 и 2.11.
|
|
Рис. 2.10. Изменение буферности в ходе титрования смеси сильной и слабой кислот сильным основанием |
Рис. 11. Кривая титрования смеси сильной и кислот сильным основанием |
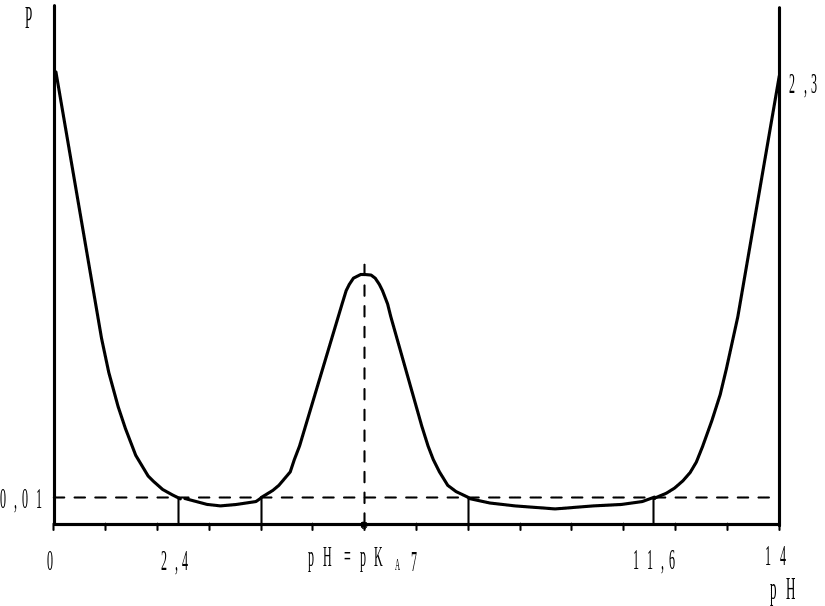
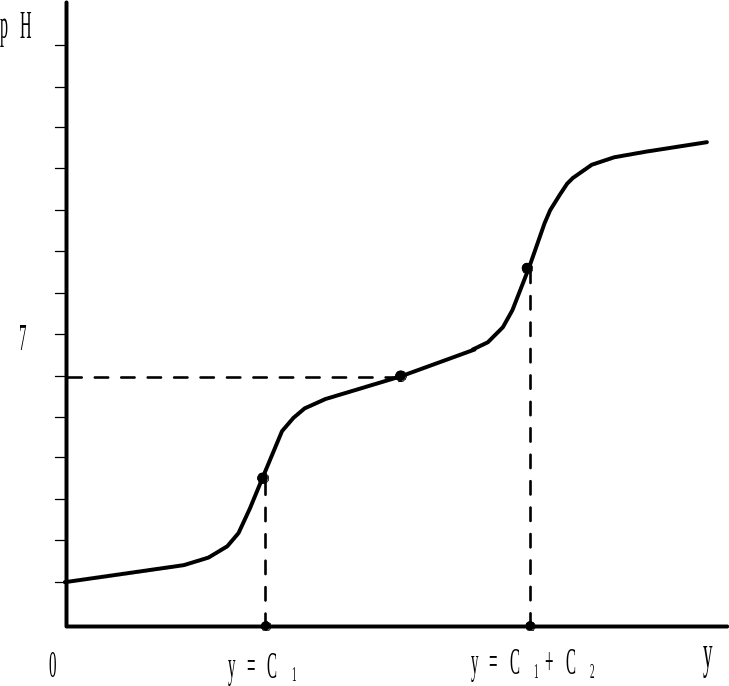
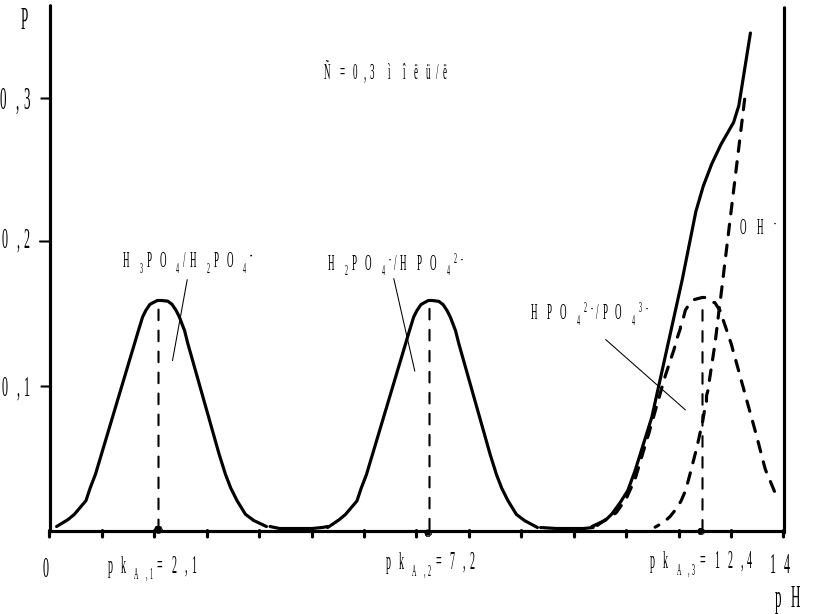
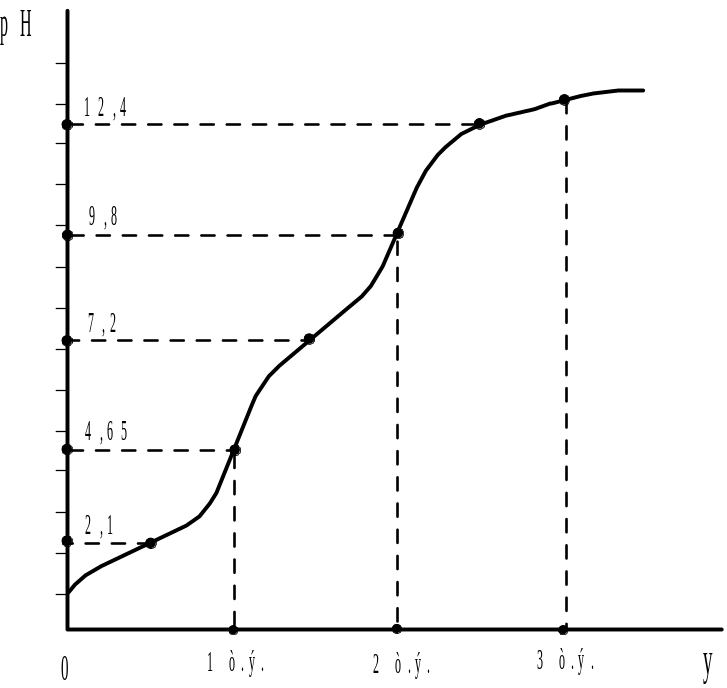
Многоосновную кислоту можно рассматривать как смесь двух или большего числа слабых кислот с идеально одинаковыми концентрациями. Поэтому при титровании многоосновных кислот остаются в силе уравнения (2.34) — (2.45), только константы диссоциации кислот КА смеси следует заменить константами ступенчатого равновесия многоосновных кислот kA , т.е. рКА,1 следует заменить показателем pkA,1, рKА,2 − показателем рkA,2 и рКА,3 − показателем pkA,3. В качестве примера на рис. 2.12 представлена кривая изменения буферности, а на рис. 2.13 кривая титрования фосфорной кислоты (с0 = 0,3 моль/л).
|
|
Рис. 2.12. Изменение буферности водного раствора фосфорной кислоты (с=0,3 моль/л) в ходе титрования сильным основанием |
Рис.2.13. Кривая титрования фосфорной кислоты сильным основанием |
Последовательные стадии диссоциации фосфорной кислоты имеют свои константы равновесия:
H3PO4 = H+ + H2PO4-, kA,1 = 7,5∙10-3, pkA,1 = 2,1;
H2PO4- = H+ + HPO42-, kA,2 = 6,3∙10-8, рkA,2 = 7,2;
HPO42- = H+ + PO43-, kA,3 = 4,0∙10-13, pkA,3 = 12,4.
В соответствии с (36) уравнение буферности трехосновной фосфорной кислоты должно иметь вид
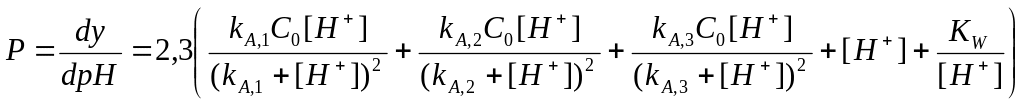 ,
(2.52)
,
(2.52)
а кривая, рассчитанная по этому уравнению и представленная на рис. 12, имеет две точки максимума, отвечающие условию рН=pkA,1 и pH=pkA,2. Третий участок высокой буферности, отмеченный пунктиром и обусловленный системой HPO42-/PO43-, перекрывается участком повышенной буферности, вызванной появлением избытка титранта − ионов ОН-. В связи с этим на кривой титрования наблюдается только два скачка. Первый из них наблюдается у первой точки стехиометричности, когда нейтрализован первый протон из трех. Так как рkA,2 - pkA,1 =5,1, то погрешность определения кислоты по первому скачку, согласно уравнения (44), будет составлять 0,29%. рН первой точки эквивалентности рН1т.э.=1/2(2,1+7,2)=4,65.
У второй точки стехиометричности также имеется скачок, практически одинаковый с первым, т.к. pkA,3 - рkA,2 =5,2. Ориентируясь на вторую точку эквивалентности, для которой рН2т.э.= 1/2(7,2+12,4)=9,8 можно определить концентрацию H3PO4 c погрешностью 0,25%. У третьей точки стехиометричности скачка нет, поскольку pkA,3>9,6. Таким образом, концентрацию фосфорной кислоты с одинаковым успехом можно определить используя первый или второй скачок на кривой титрования.
На практике при анализе многоосновных кислот встречаются самые разнообразные случаи. Например, на кривых титрования двухосновных винной (pkA,1=2,98; pkA,2=4,34) или щавелевой (pkA,1=1,23; pkA,2=4,12), а также трехосновной лимонной кислоты (pkA,1=3,1; pkA,2=4,75; pkA,3=5,4) имеется только один скачок, соответствующий переходу к средней соли. На кривой титрования двухосновной угольной кислоты (pkA,1=6,37; pkA,2=10,25), также имеется только один скачок, который соответствует переходу кислоты в кислую соль. Поэтому при титровании многопротонных кислот, чтобы не допустить ошибок в расчете концентраций и выбрать оптимальные условия анализа, необходимо знать ступенчатые константы диссоциации.
В заключение необходимо отметить, что при расчете кривых титрования оснований кислотами, общий подход остается тем же, что и для слабых кислот, но удобнее вместо рН использовать величину рОН.