

3.3. Кулонометрия
3.3.1. Классификация кулометрических методов
Кулонометрия основана на проведении электролиза исследуемого раствора, определении количества прошедшего электричества и последующем расчете количества определяемых веществ на основе закона Фарадея.
Количество прошедшего электричества можно измерить, подключая кулонометр последовательно с электрохимической ячейкой, в которой проводят электролиз. Чаще для определения Q измеряют силу тока. Поскольку мгновенное значение тока равно dQ/dt, то количество электричества равно интегралу тока по времени:
![]() .
(3.6)
.
(3.6)
С помощью этой формулы можно оценить и чувствительность метода. Учитывая, что современные приборы позволяют измерять токи на уровне 10-9 А и ниже, кулонометрическим методом можно определять нанограммовые количества вещества.
В зависимости от происходящих в растворе электрохимических процессов различают прямую кулонометрию и косвенную кулонометрию (кулонометрическое титрование).
Когда на рабочем электроде происходит одна из реакций:
A + ne- B (электровосстановление)
или
B - ne- A (электроокисление)
то, измеряя необходимое для завершения этих реакций количество электричества и зная n и M реагирующих веществ, можно определить их содержание в растворе. Этот способ относится к прямой кулонометрии.
Если вещества А и В электрохимически не активны, то в раствор можно внести такое вещество, которое способно в соответствующих условиях окисляться или восстанавливаться на электроде согласно уравнениям:
D - me- Z или Z + me- D
Продукт этих реакций (Z или D) способен химически количественно взаимодействовать с определяемым веществом В или А. Тогда количество электричества, затраченное на образование продукта Z или D, эквивалентно содержанию В или А в растворе, и оказывается возможным вычислить количество последних. Этот способ называется косвенной кулонометрией.
По технике выполнения кулонометрический метод анализа делится на потенциостатическую кулонометрию и гальваностатическую кулонометрию в зависимости от того, проводят ли электрохимические превращения веществ соответственно при постоянстве потенциала рабочего электрода или при постоянстве силы тока электролиза.
Для всех методов кулонометрии обязательным является условие, при котором превращение вещества на электроде должно протекать со 100%-ным выходом по току. Поэтому следует подбирать такие условия электролиза (рН раствора, материал электрода, растворитель, фоновый электролит и т.п.), чтобы побочные электродные реакции отсутствовали, и выход по току был близок к 100 %.
Кроме того момент завершения электрохимической реакции при прямой кулонометрии и химической реакции при косвенной кулонометрии должен быть достаточно точно устанавливаемым.
3.3.2. Прямая потенциостатическая кулонометрия
Метод основан на контроле за потенциалом рабочего электрода и на поддержании его постоянного значения в течение всего электролиза. Вследствие расхода определяемого компонента при прохождении электрического тока через ячейку его концентрация в объеме раствора непрерывно уменьшается. Это видно из серии вольтамперограмм, полученных в различное время (рис. 3.5).
Рассмотрим электролиз вещества Ох с начальной концентрацией С0 при потенциале предельного диффузионного тока Еc, протекающий по схеме
Ох + ne → Red.
В условиях потенциостатической кулонометрии при интенсивном перемешивании ток электролиза (I) в любой момент времени определяется выражением:
![]() ,
(3.7)
,
(3.7)
где D – коэффициент диффузии Ох; с – концентрация Ох; δ – толщина диффузионного слоя; А – площадь электрода.
I
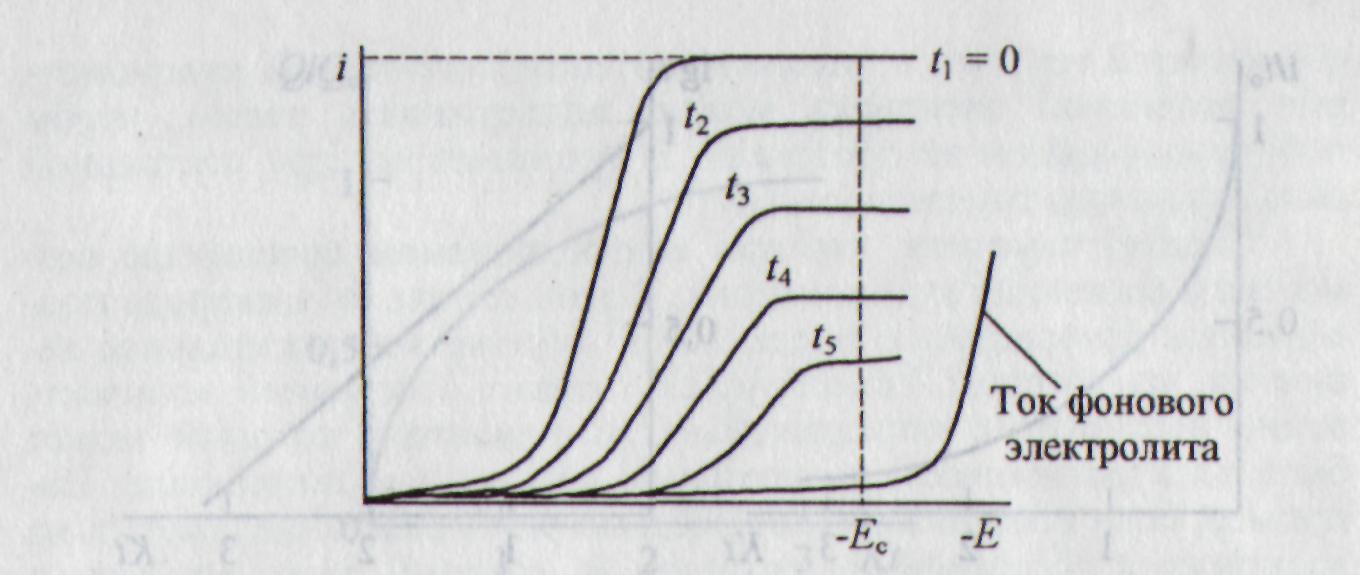
Рис. 3.5. Кривые ток-потенциал раствора вещества Ох, подвергаемого электролизу различной продолжительности
Величина I и скорость превращения dN/dt вещества Ох, связаны выражением
I = - nF[dN/dt] , (3.8)
где N - число молей вещества Ох в растворе.
Если допустить, что концентрация Ох в любой точке раствора одна и та же (например, за счет перемешивания), то в любой момент времени
с=N/V, (3.9)
где V- объем раствора.
Объединяя соотношения(3.8) и (3.9), получим
I = - nFV[dс/dt]. (3.10)
С учетом выражений (3.7) и (3.10) имеем
![]() .
(3.11)
.
(3.11)
Это уравнение можно проинтегрировать и записать в виде
![]() (3.12)
(3.12)
с начальным условием с = с0 при t = 0.
Используя выражение (3.12), получим зависимость тока от времени электролиза:
I = I0exp(-Kt), (3.13)
где I0 - ток в начальный момент электролиза; К = DA/Vδ.
Таким образом, зависимость между током электролиза и временем подчиняется кинетическому уравнению реакции первого порядка. При этом константа К зависит от коэффициента диффузии электроактивного вещества, площади электрода, объема раствора и толщины диффузионного слоя. По мере электролиза концентрация электроактивного вещества и соответствующий ей ток уменьшаются по экспоненте до уровня остаточного тока. На рис. 3.6 приведены зависимости I от времени электролиза в безразмерных координатах.
Для увеличения скорости анализа (т.е. увеличения К) целесообразно применять большие электроды и уменьшать объем испытуемого раствора. Сокращению продолжительности анализа способствует и увеличение скорости перемешивания раствора, так как при этом уменьшается , возрастает скорость диффузии и перенос вещества к диффузионному слою конвекцией.
I/I0
|
lgI
|
а |
б |
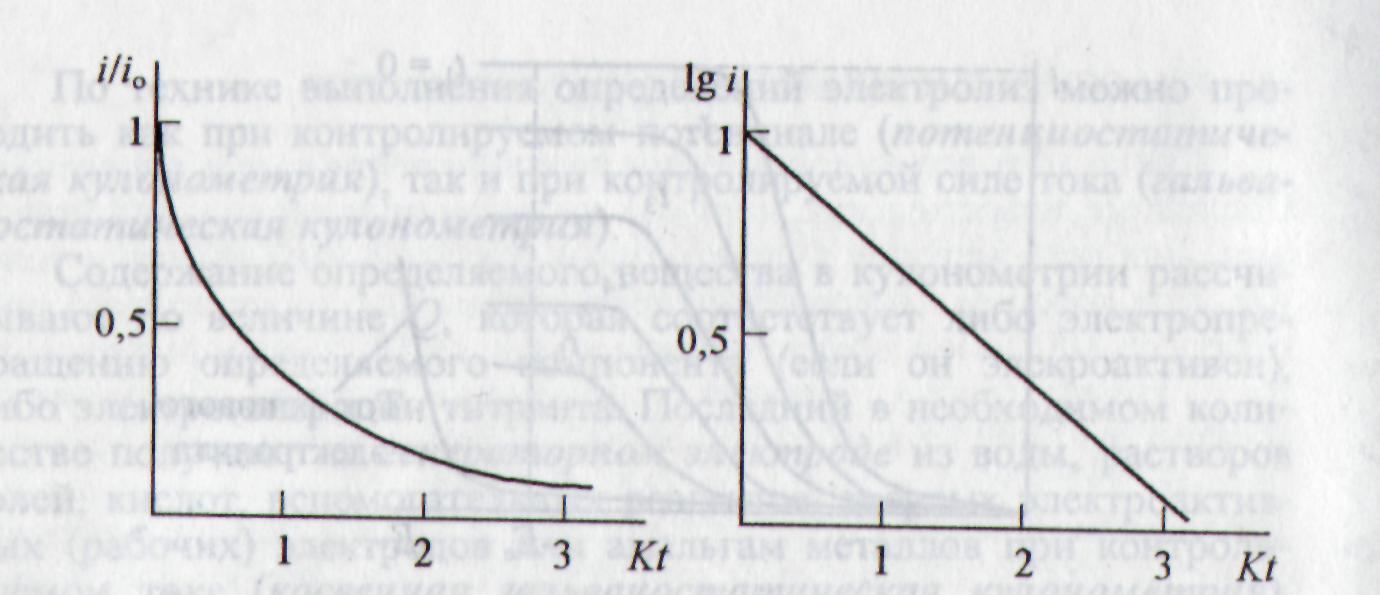
Рис. 3.6. Зависимости тока от времени электролиза (в безразмерных координатах)
При перемешивании раствора с постоянной скоростью величина предельного диффузионного тока пропорциональна концентрации деполяризатора. Чтобы подавить миграционный ток, обусловленный передвижением ионов к поверхности электродов под действием градиента потенциала, и повысить электрическую проводимость в анализируемый раствор вводят большую концентрацию электрохимически индеферентного (ионы которого не способны к восстановлению или окислению на электродах) электролита, называемого электрохимическим фоном.
Из уравнения (10) следует, что теоретически окончание электролиза (с=0, I=0) может быть достигнуто только через бесконечное время. В этом случае количество электричества можно рассчитать по формуле
![]() .
(3.14)
.
(3.14)
В действительности даже при бесконечном продолжении электролиза сила тока по достижении некоторого малого значения перестает далее уменьшаться и остается постоянной. Этот остаточный ток вызван электролизом разных микропримесей - деполяризаторов, например, восстановлением растворенного кислорода, ионов водорода и другими причинами. Поэтому электролиз следует считать оконченным в момент, когда сила тока перестает изменяться в течение некоторого времени. Однако в этом случае продолжительность анализа будет большой, а момент окончания электролиза - неопределенным, что может привести к существенным ошибкам.
Если руководствоваться заданной точностью результатов анализа, то нет необходимости продолжать процесс до прекращения изменения силы тока в цепи. Логарифмируя уравнение (3.13), получим
lg I= lg I0 – Kt/2,303. (3.15)
На основании наблюдаемой линейной зависимости lgI - t (рис. 3.6, б) легко определить постоянную К и истинное значение начального тока I0. Зная величину К, можно оценить время электролиза tэ. При степени завершенности электролиза 99% отношение c/cо=I/I0=10-2, tэ = 4,606K; для 99,9%-ной завершенности tэ = 6,909K. Такой расчет применяется для грубых оценок, потому что фактически зависимость между током и временем не следует в точности уравнению первого порядка ввиду наличия фоновых токов, эффектов заряжения и непостоянства условий переноса вещества к электроду.
Из уравнения (3.11) следует, что скорость электролиза не зависит от начальной концентрации c0 и электролиз разбавленных растворов вещества Ох до степени завершенности 99,9% при одних и тех же значениях А, V и К потребует таких же затрат времени, что и электролиз более концентрированных растворов. На рис. 3.7 приведена зависимость количества электричества от времени электролиза по мере его завершения. Практически электролиз заканчивают, когда ток снижается до величины 0,001 от первоначального значения i0. В этом случае погрешность определения не превышает 0,1%.
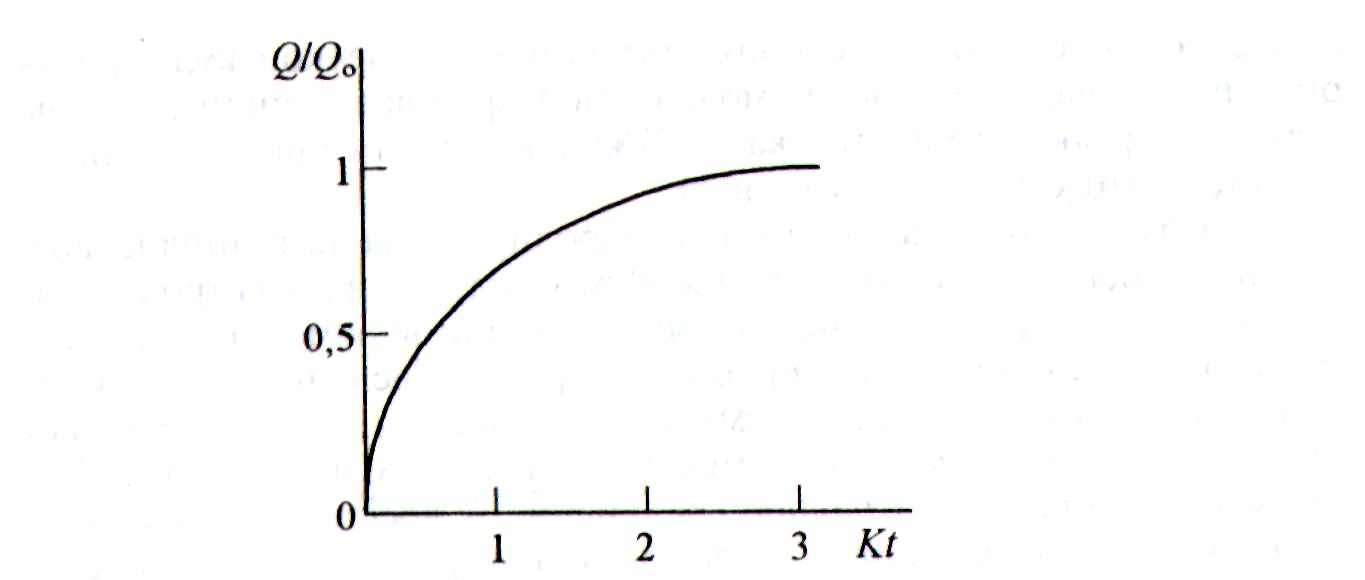
Когда электролиз проводят практически до достижения остаточного тока, при заметной величине последнего следует внести поправку в полученное значение Q, вычитая из него количество электричества Qост, равное произведению величины остаточного тока iост. на продолжительность электролиза tэ
Q = Qобщ −Qост., (3.16)
где Qобщ - количество электричества, измеренное в процессе электролиза; Qост - количество электричества, расходуемое в отсутствие определяемого вещества.
При определении больших количеств вещества Qобщ >> Qост . В этом случае точность метода зависит от точности измерения Q. Если Qост нельзя пренебречь, то точность определения зависит от воспроизводимости этой величины. В настоящее время достигается точность измерений в пределах 0,002-0,05 %. Причем концентрация вещества, установленная с помощью потенциостатической кулонометрии, ближе к его истинной концентрации в растворе, чем в случае кулонометрии с контролируемой силой тока. Это объясняется тем, что в условиях потенциостатической кулонометрии потенциал электрода можно поддерживать таким, чтобы побочные реакции не протекали. В кулонометрии при постоянной силе тока этого трудно достигнуть.
Для ускоренного определения общего количества электричества без проведения полного электролитического превращения анализируемого вещества Мейтс предложил следующую расчетную формулу:
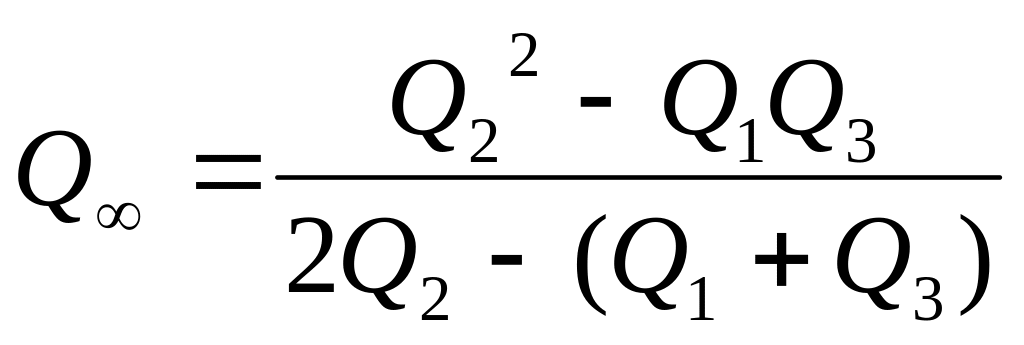 ,
(3.17)
,
(3.17)
где Q - полное количество электричества, необходимое для разложения анализируемого вещества; Q1, Q2, Q3 - количество электричества, измеренные в моменты времени t1, t2, t3 от начала электролиза, причем (t2 - t1) = (t3 - t2).
Расчет по методу Мейтса упрощается при Q1 = 0, т.е. при замере от начала электролиза. При проведении электролиза на 3/4 ошибка вычислений Q не превышает 0,7 %, что является удовлетворительным. Имеющиеся экспериментальные данные показывают, что метод потенциостатической кулонометрии удобно применять главным образом для определения миллиграмовых количеств (5 - 200 мг). Возможно приложение его и к микрограммовым количествам. Ошибка определения составляет 0,01 - 3,0 % и зависит главным образом от применяемой аппаратуры, чем от интервала измеряемых концентраций.
Избирательность определения зависит от выбора потенциала электрода, при котором протекает электрохимическая реакция в присутствии мешающих веществ. Ее можно повысить, поддерживая потенциал электрода с высокой точностью. Оптимальные условия проведения электролиза определяют из кривых ток-потенциал.
Допустим на рабочем электроде протекает реакция восстановления и в растворе присутствует три способных к восстановлению вещества А(1), А(2) и А(3), одно из которых А(2) подлежит определению. Для создания возможно большего тока потенциал электрода Ес выбирают таким, чтобы он соответствовал области предельного диффузионного тока анализируемого вещества. Согласно поляризационной кривой, снятой для этой системы (рис. 4), в качестве рабочего потенциала следует выбрать потенциал Е(2), при котором ток восстановления определяемого компонента будет максимальным. Однако при этом значении потенциала будет восстанавливаться также вещество А(1) - мешающий компонент. Перед проведением анализа его следует предварительно удалить, проведя электролиз при рабочем потенциале Е(1). Затем задать потенциал рабочего электрода Е(2) и определить количество электричества для его восстановления.
Одним из эффективных средств нивелирования влияния мешающих веществ является их связывание в прочные комплексы. Это приводит к уменьшению равновесных концентраций примесей настолько, что их потенциалы электропревращения резко смещаются в нужную сторону (в приведенном примере в катодную область) и присутствие этих соединений не влияет на результат анализа. Нередко можно также достигнуть положительных результатов изменением природы рабочего электрода, кислотности среды и некоторых других факторов.
I
|
Рис. 3.8. Вольтамперограмма раствора, содержащего три способных к восста-новлению вещества: А(2) − определяемый компонент; А(1)− мешающий компонент |

Следует заметить, что при контролируемом потенциале возможна и косвенная кулонометрия. В этом случае на электроде подвергается превращению введенное в избытке вспомогательное вещество, из которого генерируется титрант. Последний количественно реагирует с определяемым компонентом, который может быть электрохимически неактивным при данном потенциале. Например, если вспомогательное соединение А восстанавливается на электроде с образованием титранта В, который далее вступает в реакцию с определяемым компонентом X, т.е.
А + ne- → В, В + X → Р,
то электролиз будет проводиться до тех пор, пока X полностью не израсходуется. Однако косвенная кулонометрия с генерированием титранта на практике применяется в основном в гальваностатическом варианте.
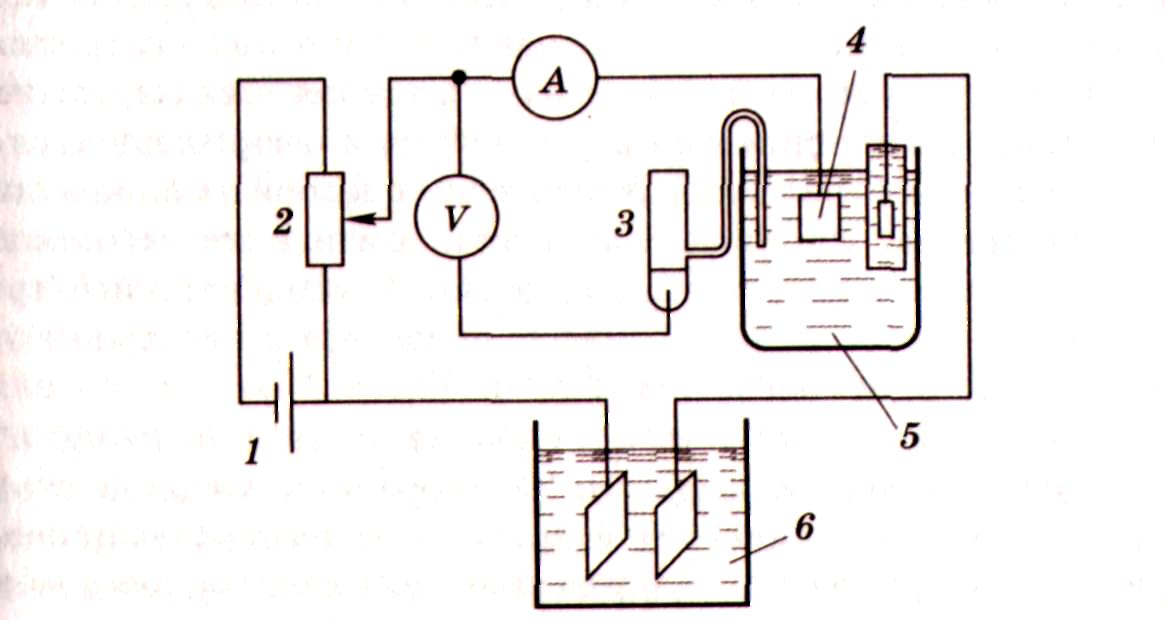
Кулонометрические методы при постоянном контролируемом потенциале широко применяются в прямой кулонометрии. Принципиальная схема установки для потенциостатической кулонометрии приведена на рис. 3.9. Напряжение с аккумуляторной батареи 1 через делитель напряжения 2 подается на рабочий электрод 4 кулонометрической ячейки 5. Потенциал электрода определяется милливольтметром или потенциометром, сила тока — амперметром. Количество израсходованного электричества измеряется кулонометром 6.
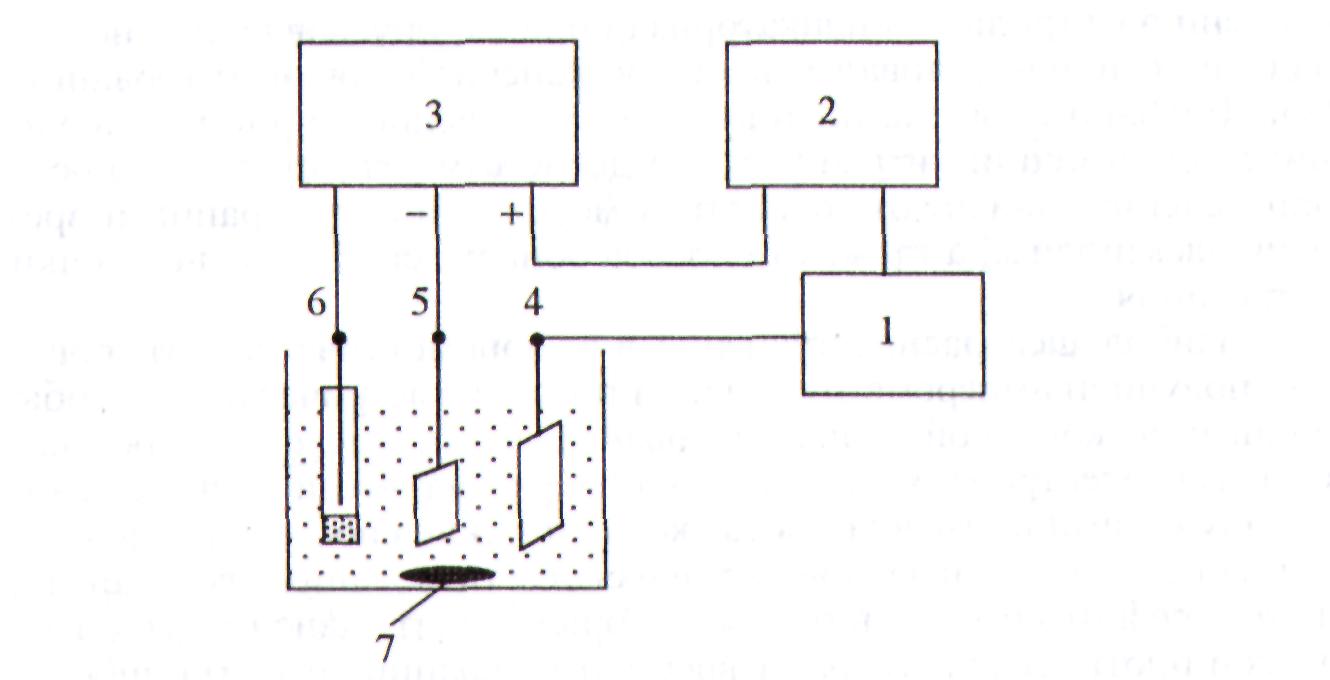
В современных установках для проведения электролиза обычно используют трехэлектродную ячейку и специальные электронные приборы — потенциостаты, поддерживающие заданный потенциал с точностью примерно ±10 мВ в интервале от -2,5 до 2,5 В (рис. 3.10). Потенциал рабочего электрода устанавливают с помощью поляризационной кривой (I—V-кривой) в области, где достигается предельный ток.
|
|
Рис. 3.9. Схема простейшей установки для потенциостатической кулонометрии |
Рис.3.10. Блок-схема установки для кулономе-трических измерений при контролируемом потенциале 1 - кулонометр; 2 -регистратор; 3 - потенциостат; 4 - вспомогательный электрод; 5 - рабочий электрод; 6 - электрод сравнения; 7 - магнитная мешалка |
Рабочим электродом кулонометрической ячейки обычно служит платиновая пластинка или ртуть, хотя иногда используют также золотые, серебряные или графитовые электроды. Вспомогательный электрод изготовляется из тех же материалов. Электродные пространства рабочего и вспомогательного электродов разделены. Контакт между ними осуществляется через пористую перегородку. В качестве электрода сравнения обычно выбирают каломельный или хлорсеребряный. Количество электричества, израсходованное на протекание электрохимической реакции, измеряется с помощью интеграторов тока или кулонометров, а также может быть определено расчетным методом.
Принцип действия кулонометров основан на том, что через последовательно включенный прибор в цепи протекает такой же ток, какой проходит через анализируемый раствор, и, следовательно, за некоторый промежуток времени через анализируемый раствор и через прибор пройдет одно и то же количество электричества. В последовательно включенном кулонометре со 100%-м выходом протекает хорошо известная электрохимическая реакция, и измерение количества электричества сводится, таким образом, к определению количества вещества, полученного в результате этого процесса.
В зависимости от способа измерения объема или массы вещества различают газовые, электрогравиметрические, титрационные и другие кулонометры. В газовых кулонометрах определяется объем газа, выделившегося в результате электрохимического процесса. В электрогравиметрических кулонометрах определяется масса вещества. Например, в медных кулонометрах находят массу металлической меди, выделившейся при электролизе сульфата меди, в серебряных — массу серебра, полученного при электролизе нитрата серебра, и т. д.