

3.3.3. Прямая амперостатическая кулонометрия
Если анализируемое вещество находится на электроде в твердом состоянии в виде металлической пленки, пленки оксида металла или соли, то для определения его количества удобно использовать гальваностатическую кулонометрию.
Так, для реакции окисления металла Ме
Me - ne- Men+
окисление можно производить при неизменяющейся величине тока I. В конце реакции, когда окислятся последние следы металла, электроны поставляются за счет окисления молекул воды (в случае отсутствия других веществ, способных к окислению в условиях электролиза):
2.H2O - 4e- O2 + 4H+ (в кислой среде)
4OH- - 4e- O2 + 2H2O (в щелочной среде)
Изменение природы электродной реакции будет сопровождаться резким изменением потенциала, что позволяет надежно зарегистрировать время и количество электричества, затраченное на окисление металла, а затем рассчитать количество окисляющегося металла. Основной недостаток метода состоит в том, что последние следы вещества сходят с поверхности электрода неравномерно, вызывая значительные изменения величины тока, которые не поддаются регулированию.
Аналогичные явления происходят при восстановлении оксидов металла до металла, которое часто протекает с промежуточным образованием низшего оксида, а после его восстановления до металла начинается восстановление молекул воды. Каждой стадии восстановления соответствуют скачки потенциала рабочего электрода.
Метод можно использовать также для определения веществ, присутствующих в растворе и для предварительного выделения анализируемого вещества на твердом электроде (концентрирования) с последующим растворением (инверсионная кулонометрия).
Так же, как и в потенциостатической кулонометрии, ход электролиза при контролируемой силе тока (Ic) можно проиллюстрировать с помощью серии кривых ток-потенциал (рис. 7). Поскольку в начале электролиза ток Iс меньше, чем предельный диффузионный ток для соответствующей концентрации электроактивного вещества, то электродная реакция протекает со 100%-ным выходом по току. По мере электролиза объемная концентрация Ох уменьшается вместе с Iпр . В случае, когда
с = Ic /nFKV, (3.18)
величина Iс равна Iпр (кривая 5). В ходе дальнейшего электролиза Iпр становится меньше Iс, и потенциал электрода сдвигается к более отрицательным значениям (рис. 8, a), при которых может протекать другая электрохимическая реакция. Ток этой реакции будет равен Iс. В этом случае выход по току станет меньше 100 %. Поэтому электролиз прекращают, когда наблюдается скачок потенциала. По перегибу кривой зависимости Е = f(t) можно найти время завершения электрохимической реакции.
I
Ic
|
|
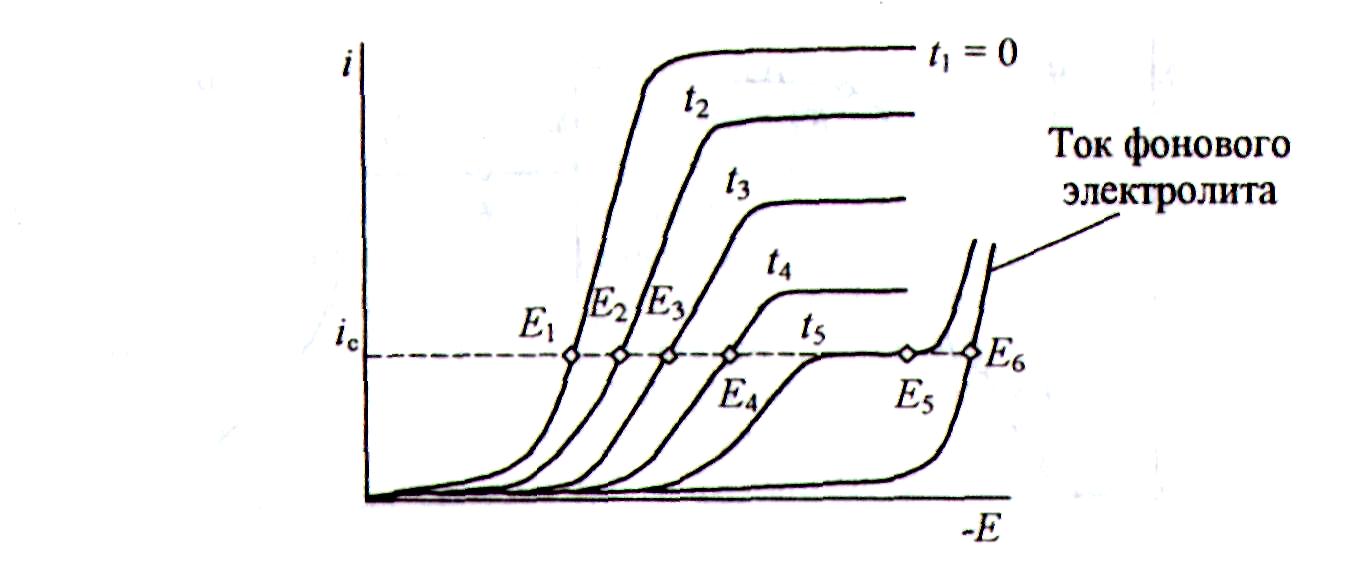
Рис. 3.7. Изменение кривых ток-потенциал в зависимости от времени электролиза при контролируемой силе тока
Очевидно, что кулонометрия при контролируемой силе тока имеет меньшую селективность, чем кулонометрия при контролируемом потенциале, поскольку в определенный момент времени (Iпр < Ic) может пойти реакция с участием мешающего вещества, фонового электролита или растворителя. При этом скорость электролиза по существу будет равна скорости превращения вещества Ох при контролируемом потенциале, но с заметно меньшим выходом по току, который начинает уменьшаться по экспоненциальному закону (рис. 8, б).
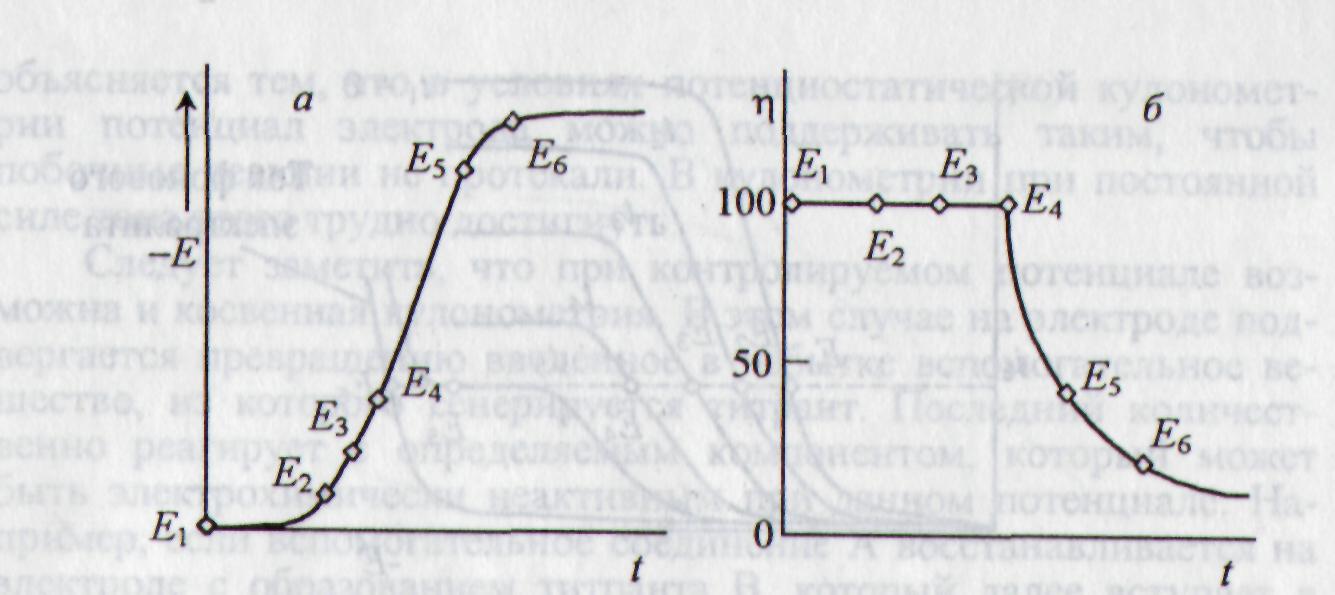
Рис. 8. Изменение потенциала (а) и выхода по току (б) со временем в условиях электролиза при контролируемом токе
Величину тока в гальваностатической кулонометрии поддерживают постоянной с точностью до ± 0,1 %. Продолжительность электролиза измеряют с помощью хронометра и рассчитывают количество электричества, прошедшее через ячейку: Q = Ic∙t. Короткие промежутки времени измеряют с помощью осциллографов или электронных секундомеров с точностью до 0,001 с. Современные приборы имеют достаточную точность для того, чтобы этот фактор не учитывать.
Метод позволяет определять очень малые количества вещества с большой точностью. При токе электролиза 10-6 А возможно определение 10-11 моль/л вещества, что эквивалентно приблизительно 10-9 г. Однако правильность определений ограничена отклонением значений выхода по току от 100 % при Iпр < Ic . Добиться 100 %-ного выделения какого-либо вещества очень трудно, так как на электроде должно окисляться или восстанавливаться только одно это вещество.