

2. Потенциометрия
Потенциометрия объединяет методы определения различных физико-химических величин и концентрации веществ, основанные на измерении электродвижущей силы (ЭДС) обратимых электрохимических цепей, когда индикаторный (рабочий) электрод имеет потенциал весьма близкий к равновесному значению. Поэтому потенциометрия это равновесный электрохимический метод анализа.
Основы потенциометрии заложены В. Нернстом, который в 1889 г. получил известное уравнение для равновесных электродных потенциалов. Вскоре потенциометрия начала применяться в аналитической химии, а в 1893 г. Р. Беленд провел первое потенциометрическое титрование. С этого времени в зависимости от целей измерения потенциала различают два вида потенциометрии: косвенная потенциометрия (потенциометрическое титрование) и прямая потенциометрия (ионометрия).
2.1. Потенциометрическое титрование
2.1.1. Индикаторные электроды, преимущества потенциометрической индикации конечной точки титрования
Потенциометрический метод титрования, является разновидностью объемного анализа и основан на определении конечной точки титрования (КТТ) по результатам измерения потенциала индикаторного электрода, реагирующего на изменение активности одного из участвующих в реакции веществ или продукта реакции.
По сравнению с визуальной индикацией КТТ потенциометрический метод имеет ряд преимуществ:
как инструментальный метод исключает субъективные ошибки;
позволяет осуществлять титрование в мутных и окрашенных растворах;
дает возможность дифференцировано определять компоненты смесей из одной порции раствора;
легко поддается автоматизации.
В потенциометрии могут быть использованы все четыре типа химических реакций: кислотно-основные, окисления-восстановления, осаждения и комплексообразования. К химическим реакциям в потенциометрии предъявляют обычные для объемного анализа требования: большая скорость, количественное протекание в нужном направлении (большая константа равновесия), строгая стехиометричность и отсутствие побочных реакций. Кроме того для проведения анализа необходимо иметь индикаторный электрод, обратимый относительно одного из участников реакции.
Выбор индикаторного электрода определяется типом реакций и их конкретными участниками. Наиболее просто подобрать рабочий электрод для реакций окисления-восстановления и кислотно-основных.
В первом случае универсальным электродом является платиновый электрод, с помощью которого независимо от природы конкретного окислителя или восстановителя можно зарегистрировать изменение окислительно-восстановительного потенциала в ходе титрования и тем самым найти конечную точку титрования.
В кислотно-основном титровании независимо от природы определяемых кислот или оснований необходимо зарегистрировать величину или изменение концентрации ионов водорода, поэтому в качестве индикаторного можно использовать любой электрод, функционирующий как водородный, т.е. обратимый относительно ионов H+. К таковым относятся газовый водородный, хингидронный, металл-оксидные (например, сурьмяный) и стеклянный электроды.
Индикаторные электроды в реакциях осаждения и комплексообразования являются избирательными. Это объясняется тем, что виды ионов, входящих в состав осадков и комплексов, самые разнообразные, а индикаторный электрод должен быть обратимым относительно одного из вида.
Это условие трудно выполнить из-за большой электролитической упругости растворения большинства металлов либо по другим причинам. Из электродов 1-го рода в качестве индикаторных в водных средах используются медный, ртутный, серебряный, потенциалы которых имеют более положительное значение, чем потенциал водородного электрода. Применение электродов из металлов, стоящих в ряду напряжений левее водорода, исключено, т.к. они являются необратимыми ввиду восстановления на них ионов водорода (или молекул воды в щелочной среде). Это ограничивает использование потенциометрического метода для реакций осаждения и комплексообразования. Для таких реакций в качестве индикаторных электродов часто используются электроды 2-го рода (например, хлорсеребряный, ртутно-сульфатный и др.), 3-го рода (примером может служить ртутный электрод, опущенный в насыщенный раствор Hg2C2O4 и CaC2O4, содержащий в избытке определяемые ионы Ca2+), а также амальгамные (например, амальгама кадмия для определения ионов Cd2+) и ионоселективные электроды.
Потенциометрическое титрование проводят следующим образом: в титруемый раствор помещают индикаторный электрод и электрод сравнения, потенциал которого в ходе титрования остается постоянным. Иногда электрод сравнения помещают в отдельный сосуд, который соединяют с сосудом для титрования при помощи солевого мостика. Система из индикаторного электрода и электрода сравнения образует гальванический элемент, который подключают к потенциометрической установке для измерения ЭДС.
К раствору анализируемого вещества из бюретки прибавляют раствор взаимодействующего с ним реагента. При ручном титровании прибавление реагента ведут определенными дозами. Используя автоматическую бюретку, подачу титрующего реагента можно осуществлять непрерывно с определенной скоростью или дискретно с заданным объемом титранта. Зависимость измеренной в ходе титрования ЭДС от объема добавленного раствора титранта изображают графически. Такую зависимость называют кривой потенциометрического титрования.
Кривая потенциометрического титрования может быть разделена на несколько участков, отличающихся темпом изменения потенциала на единицу объема добавленного титранта. Начальный участок кривой (примерно до приливания 90 % всего теоретически необходимого количества титранта) характеризуется медленным изменением потенциала.
В точке эквивалентности (ТЭ) количество реагента, добавленного в титруемый раствор, строго эквивалентно количеству определяемого вещества. Вблизи ТЭ незначительное изменение количества введенного титранта вызывает резкое изменение активности потенциалопределяющих ионов и соответствующее изменение потенциала индикаторного электрода. В практике используют термин «скачок титрования», означающий изменение потенциала индикаторного электрода при изменении степени оттитрованности раствора от 99,9 до 100,1 %. При дальнейшем изменении объема титранта после ТЭ скорость изменения потенциала замедляется. Титрант прибавляют до получения значений индикаторного электрода, практически не изменяющихся при дальнейшем титровании. Величина скачка титрования связана с константой соответствующего химического равновесия и с условиями определения (концентрации титруемого раствора и титранта, температура и т.д.).
Независимо от техники измерения ЭДС (компенсационным методом, современным рН-метром или цифровым вольтметром), наиболее распространенным приемом определения КТТ в потенциометрии является графический способ. Он заключается в построении интегральной кривой титрования (рис. 3.1а) в координатах ЭДС гальванического элемента Е (или потенциал индикаторного электрода) – объем прибавленного раствора титранта V (или количество титранта y).
Точка перегиба (точка, в которой кривая титрования имеет максимальную крутизну в момент “скачка” потенциала) на кривой титрования E=f(V) принимается обычно за КТТ, которая может совпадать или не совпадать с ТЭ, но должна быть достаточно близкой к ней и обеспечивать необходимую точность анализа. Несоответствие координат КТТ и ТЭ возникает в тех случаях, когда определяемые ионы и ионы титранта имеют различные заряды, т.е. стехиометрия реакции отличается от соотношения 1:1. Точка максимального наклона S-образной кривой находится с той стороны от точки эквивалентности, где в избытке присутствует ион с меньшим зарядом. Правильность нахождения ТЭ определяется константой равновесия протекающей при титровании химической реакции и быстротой достижения равновесия химической и электродной реакции. При уменьшении этих параметров ошибка титрования возрастает.
Для нахождения точки перегиба обычно проводят две параллельные касательные к пологим нижней и верхней ветвям кривой и соединяют их прямой так, чтобы точка пересечения ее с кривой титрования делила эту прямую на две равные части (точка А, рис. 2.1г).
Имеются и другие геометрические способы нахождения точки перегиба. Например, метод определения точки пересечения кривой титрования с прямой линией, соединяющей центры двух окружностей, наилучшим образом согласующихся с закругленными верхней и нижней частями кривой титрования (рис. 2.1д).
В случае, если скачок на кривой титрования размыт и определение к.т.т. затруднено (например, при малых значениях констант равновесия химических реакций), пользуются построением графической зависимости первой или второй производной потенциала от объема титранта. В первом случае КТТ соответствует максимальное значение dE/dV (рис. 2.1б), во втором – значение d2E/dV2, равное нулю (рис. 2.1в).
Интегральная и дифференциальные кривые титрования могут быть получены вручную или автоматически. В последнем случае используют автоматическую бюретку, ленточно-диаграмный самописец, блок электронного дифференцирования. Автоматические бюретки представляют собой шприцы, поршень которых приводится в движение электродвигателем. Чаще всего используются бюретки, подающие титрант с постоянной скоростью, синхронизированной с протяжкой ленты самописца. Существуют также титраторы, в которых применяются бюретки с изменяемой (регулируемой) скоростью подачи титранта и двухкоординатные самописцы. Использование автоматического титрования, исключает ошибку, неизбежную при построении интегральных и дифференциальных кривых титрования.
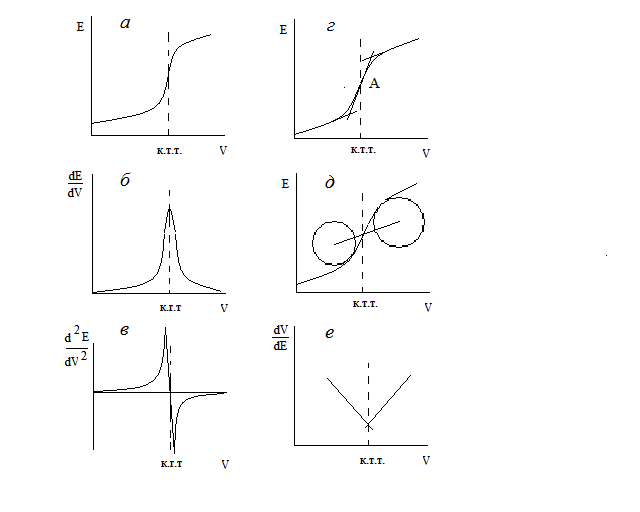
Рис. 2.1. Формы кривых потенциометрического титрования и методы определения конечной точки титровании (к.т.т.):
а – интегральная кривая; б – дифференциальная кривая по первой производной; в – дифференциальная кривая по второй производной; г – метод определения к.т.т. построением касательных; д – метод концентрических дуг; е – метод Грана.
Существует также расчетный графический метод Грана, который основан на линеаризации кривых титрования путем построения зависимости dV/dE от объема добавленного титранта V. Кривая титрования вблизи от точки эквивалентности трансформируется в две линейные ветви (рис. 2.1е), точка пересечения которых в идеальном случае совпадает с т.э.