

2. Кондуктометрические методы исследований
Теоретические основы
Кондуктометрические методы основаны на зависимости между электрической проводимостью раствора и концентрацией ионов в этом растворе. Электрическая проводимость раствора электролита — это результат диссоциации растворенного вещества и миграции ионов под действием внешнего напряжения в электрическом поле: движение ионов в растворе тормозят молекулы растворителя и окружающие, противоположно заряженные ионы. Это так называемые релаксационный и электрофоретический эффекты. Результат такого тормозящего действия — сопротивление раствора прохождению электрического тока. Электрическая проводимость раствора зависит в основном от числа, скорости (подвижности) мигрирующих ионов, количества переносимых ими зарядов, температуры и состава растворителя.
Различают удельную χ. и молярную электрические проводимости λ раствора. Удельная электрическая проводимость (См/м) — это проводимость 1 м3 раствора, находящегося между электродами площадью 1 м? каждый, расстояние между которыми равно 1 м:
χ = αСF(z+u+ + z-u-)
где α —степень диссоциации электролита; С — концентрация электролита; F — постоянная Фарадея; z+u+ и z-u--—скорости движения (м/с) и заряды катионов и анионов соответственно при напряженности электрического поля 1 В/см.
Молярная электрическая проводимость (См·м2/моль) — это электрическая проводимость раствора, содержащего 1 моль электролита, измеренная при расстоянии между электродами 1 м. Удельная и молярная электрические проводимости связаны между собой уравнением
λ = χС
При кондуктометрическом методе измеряемый аналитический сигнал — электрическая проводимость раствора. По мере повышения концентрации растворенного электролита увеличивается количество ионов-переносчиков заряда, т. е. растет χ.
Однако после достижения определенного максимума значение и начинает падать, так как в сильных электролитах усиливаются релаксационный и электрофоретический эффекты а в слабых - уменьшается степень диссоциации. Электрическая проводимость бесконечно разбавленного раствора U зависит от подвижности ионов при отсутствии тормозящих эффектов.
λ = λ∞(А
+ В)
![]()
Метод можно применять в прямой кондуктометрии или кондуктометрическом титровании. Прямую кондуктометрию используют для определения концентрации растворов довольно редко, поскольку регистрируемый аналитический сигнал не избирателен, т. е. электрическая проводимость—величина аддитивная, определяемая наличием всех ионов в растворе. Прямые кондуктометрические измерения успешно используют, например, для оценки чистоты растворителя, определения общего солевого состава морских, речных и минеральных вод, а также для определения таких важных для аналитической химии величин, как константы диссоциации электролитов, состав и константы устойчивости комплексных соединений, растворимости малорастворимых электролитов.
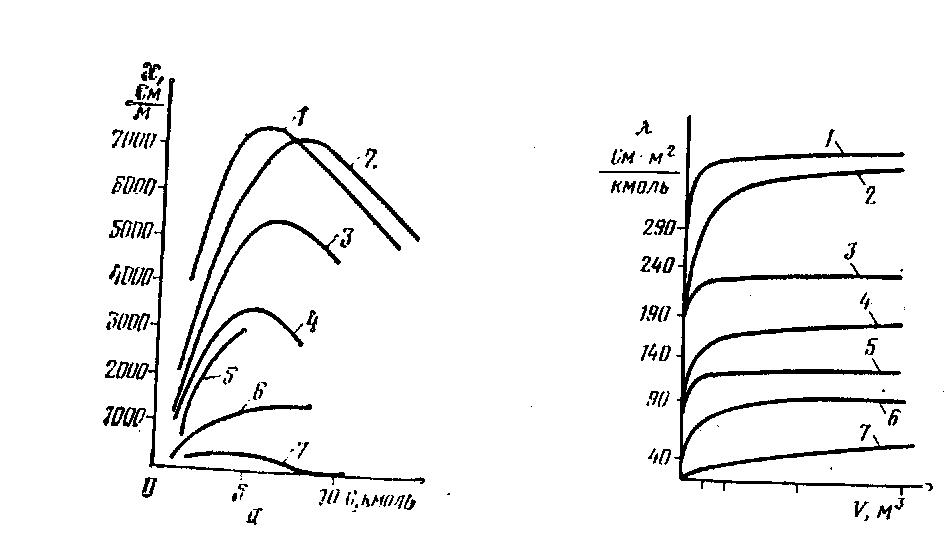
Электрическую проводимость растворов можно измерять с высокой точностью только в разбавленных растворах. В этом случае выполняются требования теории межиоиного взаимодействия Дебая—Гюккеля—Онзагера и зависимость λ = ƒ линейна для I—одновалентного электролита (зависимость λ = ƒ /υ нелинейная— см. рис. 1, б). Отклонение от линейной зависимости λ = ƒ свидетельствует об образовании ассоциатов ионных пар. В действительности линейная зависимость верна только для растворов электролитов в отсутствие примесей ионного характера. Поэтому предпочтительнее использовать для анализа метод кондуктометрического титрования, а не прямой кондуктометрии.
|
Рис. 1. Графики зависимости: а – удельной электрической зависимости от концентрации раствора электролита (С); б молярной от его разбавления (V = 1/С); 1 – HCl; H2SO4; 3 – KOH; 4 – NaOH; 5 – KCl; 6 – KCl; 7 – CH3COOH |
Метод кондуктометрического титрования основан на использовании химической реакции, в результате которой происходит заметное изменение электропроводности раствора. При таком титровании можно использовать химические реакции всех типов. Так как электрическая проводимость — функция концентрации, она должна изменяться в ходе титрования. На рисунке 2, а представлены кривые кондуктометрического титрования кислот различной силы и их смесей раствором сильного основания. По излому на кривой можно определить точки эквивалентности. Кондуктометрические кривые титрования оснований растворами НС1 представлены на рисунке 2, б.
|
Рис. 78. Кривые кондуктометрического титрования: а - кислот раствором NaOH: 1 - сильная кислота; 2 - слабая кислота; 3 - смесь сильной и слабой кислот; χ - удельная электрическая проводимость; ТЭ - точка эквивалентности; V NaОН - объем титранта; б — то же индивидуальных веществ основного характера и их смесей 1 н. раствором HCI: 1 — аммиак; 2 - анилин; 3 - ацетат натрия; 4 - едкий натр; 5 - едки натр + аммиак; 6 - едкий натр + анилин |

При кондуктометрическом титровании для получения резкого излома на кривых титрования учитывают эффект разбавления. Эффект разбавления можно свести к минимуму, титруя большой объем разбавленного раствора в ячейке концентрированным раствором из микробюретки.
Для получения надежных результатов при кондуктометрическом титровании следует иметь в виду, что величина к, изменяющаяся в процессе химической реакции, — аналитический сигнал, зависящий от многих факторов, которые надо учитывать: температуры констант образования (диссоциации) всех участников химической реакции, константы автопротолиза растворителя, подвижности ионов, ионной силы раствора и др. Использование неводных органических растворителей значительно расширяет возможности кондуктометрического метода анализа.
Правильным подбором титранта и растворителя создают благоприятные условия для титрования, при которых получают кривую с резким изломом, и погрешность определения конечной точки титрования невелика. Присутствие посторонних электролитов со значительной электропроводностью мешает определению, так как фоновый сигнал становится столь значимым, что не удается зарегистрировать изменение электропроводности в ходе титрования,
Кондуктометрическое титрование обладает рядом достоинств: оно дает возможность дифференцирование титровать смеси ряда кислот или оснований, мутные и окрашенные растворы, а также гидролизующиеся соли. Нижний предел определяемых концентраций 10-4 моль/л, погрешность определений 2%.
Электрическую проводимость раствора (или его сопротивление) измеряют в соответствующей электролитической ячейке, представляющей собой стеклянный сосуд с вмонтированными электродами. Конструкция ячейки для кондуктометрических измерений должна соответствовать интервалу измеряемых сопротивлений, и константа ячейки при этих измерениях должна оставаться постоянной. Константа ячейки (А, см-1) определяется площадью электродов (S, см2), расстоянием между ними (L, см) и зависит от формы сосуда, объема раствора и частоты тока:
A = L/S.
Прямое измерение константы ячейки невозможно, ее определяют, используя стандартные растворы КС1, для которых известны значения удельной электрической проводимости при различных температурах. Измерив сопротивление R ячейки, заполненной раствором КС1, и воспользовавшись табличным значением χ, из соотношения Л = χR вычисляют константу ячейки A.
Как правило, электроды, изготовленный из листовой платины, жестко закреплены, так что расстояние между ними не изменяется. Поэтому ячейки с определенным расстоянием между электродами и площадью поверхности пластин выбирают в зависимости от сопротивления раствора: чем выше измеряемое сопротивление, тем больше должна быть площадь электродов и меньше расстояние между ними. С учетом этого выбирают ячейку.
В агрохимических исследованиях кондуктометрию применяют для определения влажности, содержания солей в воде и почве, автоматического контроля уровня грунтовых вод при поливе, изучении динамики поступления питательных веществ к корневой системе растений. Кратко рассмотрим некоторые методы.
При кондуктометрическом методе определения влажности принцип действия различных влагомеров основан на измерении электрической проводимости растворов. Такие приборы наиболее часто используют для определения влажности зерна. Пробу зерна в измельченном виде помещают в специальный сосуд между двумя электродами и измеряют ее сопротивление. Чем больше влаги содержится в зерне, тем больше его электрическая проводимость. Шкала прибора градуирована в массовых долях (процентах влажности) для каждого вида зерна. Для разных видов сельскохозяйственных культур (пшеница, рожь, ячмень, кукуруза и др.) применяют сменные шкалы. Указанный метод отличается не только быстротой, но и довольно высокой точностью.
Влажность почвы определяют при введении специальной штанги с электродами на заданную глубину. Недостаток этого метода заключается в том, что для каждого вида почв нужно строить отдельную шкалу зависимости влажности от электрической проводимости. Кроме того, на точность определения влажности влияет засоленность почвы.
|
Рис. 3. Блок-схема установки для определения скорости растворения различных минеральных удобрений в проточном режиме: 1 стеклянный пористый фильтр с гранулами удобрений и песком; 2— насос; 3 - ячейка для измерения электрической проводимости; 4 - реохордный мост; 5 - генератор звуковой частоты тока; 6 - самопишущий потенциометр |

Более точный метод определения влажности — измерение электрической проводимости серии электродов, залитых, в гипсовые формы и постоянно находящихся в почве. Для исследования растворимости различных минеральных удобрений применяют хронокондуктометрический метод, который заключается в фиксировании перехода питательных элементов в раствор в условиях водного проточного режима. Растворение удобрений в почвенных условиях сопровождается оттоком питательных элементов за счет поглощения их растениями или движения дождевых или грунтовых вод. Несмотря на то, что процессы растворения удобрений в воде и в почве неидентичные, метод позволяет сравнивать физико-химические свойства различных удобрений, а также предварительно оценивать эффективность того или иного способа их модифицирования без продолжительных по времени агрохимических испытаний.
Сущность метода заключается в растворении навески удобрений в специальной растворительной камере в потоке растворителя, поступающего с фиксированной скоростью (рис. 3). Датчиком концентрации служат платиновые электроды, впаянные в нижней части камеры. Самопишущий потенциометр 6 через выпрямительное устройство обеспечивает автоматическую запись кондуктометрических кривых (рис. 4). Возможен последующий аналитический контроль растворов вымывания, а также качественное определение нерастворившегося остатка.
|
Рис. 3. Кондуктометрические кривые растворения различных удобрений в промывном режиме: а- миачная селитра; б - капсулнрован-ная аммиачная селитра; в — многослойное медленнодействующее удобрение
|

Промышленность выпускает реохордные мосты различных типов (Р-38, Р-556, Р-557 и другие), которые успешно используют для кондуктометрических измерений. Кондуктометр «Импульс» (позволяет измерять удельную электрическую проводимость растворов в (Интервале 1...10-6 См-м-1.
Потенциометрические методы анализа
Один из основных методов физико-химического анализа — потенциометрию — используют для определения активной концентрации элементов в тканях растения и в почве. Потенциометрия представляет собой электрохимический анализ растворов электролитов.
Развитие современных методов потенциометрического анализа идет по нескольким направлениям:
Этот метод позволяет вести измерения в мутных и окрашенных растворах, пастах и даже живых биологических объектах. Причем можно исследовать многокомпонентные смеси веществ без предварительного их разделения. Измерения выполняют очень быстро, что позволяет осуществить непрерывный контроль за процессами. При автоматизации определения ведут в широком диапазоне концентраций. Эффективный рабочий диапазон составляет 5...7 порядков, а для определения рН—около 16. Точность определений —0,1%.
|
|
Потенциометрия |
|
|
|
|
|
|
|
Редоксометрия |
|
|
|
Потенциометрическое титрование |
|
|
Ионометрия |
|
|
|
|
|
|
|
рН метрия |
|
Катионометрия |
|
Анионометрия |






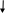
Как известно, многие соединения в растворах диссоциируют на катионы с положительным зарядом и анионы с отрицательным. Ионы и молекулы в растворе взаимодействуют друг с другом и проявляют свои свойства так, как будто концентрация их меньше, чем в действительности. Для описания явления кажущегося снижения концентрации раствора Г. Льюис в 1915 г. ввел понятие активной концентрации, или просто активности. В общем случае активность зависит от концентрации электролита. Эту зависимость выражают формулой
α = γС
где α—-• активность; С — концентрация; γ - коэффициент активности.
Активность определяют в единицах концентрации.
По определению при С→0 γ→ 1. Степень отклонения коэффициента активности от единицы служит мерой отклонения раствора от идеального состояния. Активность частиц зависит не только от электростатического, но и от химического их взаимодействия. Многие причины, приводящие к проявлению активности, известны.
П. Дебай и Э. Хюккель предположили, что в растворах сильных электролитов каждый ион окружен преимущественно противоположно заряженными ионами, или ионным облаком. Для численной характеристики ионного облака ими же было введено понятие ионной силы раствора, определяемой уравнением диффузии равны. Например, для 0,01 М растворов хлорида калия и аммония эта величина равна 1,30 мВ.
I = ½ ∑izi2
где zi и Ci - заряд и концентрация i –го иона.
Электрод, потенциал которого зависит от активности (концентрации) определенных ионов в растворе, называют индикаторным. Для измерения потенциала индикаторного электрода в раствор погружают второй электрод, потенциал которого не зависит от концентрации определяемых ионов. Такой электрод называют электродом сравнения или вспомогательным электродом.
Потенциометрический метод основан на измерении электродвижущих сил (ЭДС) гальванических элементов, включающих два электрода, которые можно погрузить в один и тот же раствор (элемент без переноса) или в два различных по составу раствора, имеющих между собой жидкостный контакт (цепь с переносом).
Рассмотрим два основных класса индикаторных электродов, используемых наиболее широко в агрохимических исследованиях.
Электроды, на межфазных границах которых протекают реакции с участием электронов, называют электронообменными, например, окислительно-восстановительные электроды.
В окислительно-восстановительных реакциях Аох + nе- = АRed присутствующие в растворе ионы в окисленной (Аox) и восстановленной (ARed) формах образуют редоксипару. Количественную зависимость электродного потенциала от концентрации окислителя и восстановителя определяют по уравнению^
E = E0 + |
0.059 |
lg |
[Ox] |
n |
[Red] |
где [Ох] — концентрация окисленной формы ионов; [Red]—концентрация восстановленной формы ионов; E0 — стандартный электродный потенциал редоксипары (синоним: окислительно-восстановительная пара).
Когда стехиометрические коэффициенты реакции перехода окисленной формы в восстановленную не равны единице, они входят в уравнение как показатели степеней соответствующих концентраций.
Коэффициент активности зависит только от концентрации электролита. Чем больше концентрация, тем сложнее эта зависимость. Обычно тип электролита обозначают двумя цифрами, первая из которых соответствует числу катионов, а вторая — числу анионов, например 1 : 1 для хлорида калия или 1 : 2 для хлорида бария. Для водных растворов электролитов состава 1 : 1 с ионной силой до 10-3 М справедлив предельный закон Дебая—Хюккеля:
lg γi = Azi2√Ī,
где А — константа, зависящая от температуры и диэлектрической проницаемости растворителя.
При большой концентрации электролитов необходимо использовать более сложные уравнения и электроды с большим избирательным потенциалом.
Электроды, на межфазных границах которых протекают ионообменные реакции, называют мембранными, или ионообменными, а также ионоселективными.
Ионоселективные электроды разделяют на следующие группы или гетерогенной мембраной; жидкостные электроды (на основе ионных ассоциатов, хелатов металлов и нейтральных лигандов); газовые электроды; электроды для измерения активности (концентрации) биологических веществ.
Характеристика электродов
Электроды сравнения. Потенциал отдельно взятого индикаторного электрода нельзя измерить. Для этого составляют гальванический элемент из измерительного электрода и электрода сравнения, потенциал которого известен.
Электроды с известным и постоянным скачком потенциала называют электродами сравнения. В качестве электродов сравнения выбирают так называемые неполяризующиеся электроды, особенность которых заключается в том, что потенциал этих электродов практически не изменяется, (они не поляризуются) при прохождении через них электрического тока. Примером таких электродов служит насыщенный хлорсеребряный электрод, который изготавливают методом электролитического нанесения хлорида серебра на серебряную проволоку. Электрод погружают в раствор хлорида калия который находиться в сосудах, связанных солевым мостиком с анализируемым раствором. Так как в концентрированных хлоридных растворах хлорид серебра растворяется с образованием хлорсеребряных комплексов, растворы хлорида калия перед погружением в них электродов обычно насыщают хлоридом серебра. При работе с хлорсеребряным электродом необходимо следить за тем, чтобы внутренний сосуд был заполнен насыщенным раствором КС1 Наша промышленность выпускает хлорсеребряные электроды марок ЭВЛ-1МЗ, ЭВЛ-1М1
|
Рис. 1. Устройство хлорсеребряного электрода ЭВЛ-1МЗ: 1 — серебряная проволока; 2 — кристаллы AgCl; 3 — резиновая пробка и отверстие для залива раствора КС1; 4 — раствор КС1; 5 — асбестовая нить; 6 — корпус; 7 — капилляр.
|

Кроме хлорсеребряного, в качестве электродов сравнения применяют каломельный, основной недостаток которого — применение токсичной ртути в составе.
Потенциал вспомогательного электрода нужно учитывать при определении окислительно-восстаноительных потенциалов в потенциометрии Для хлорсеребряного электрода потенциал при 25°С равен + 201 мВ, а каломельного +244 мВ.
Ионоселективные электроды. Ионоселективный (мембранный) электрод представляет собой гальванический полуэлемент, состоящий из ионоселективной мембраны, внутреннего раствора и внутреннего электрода сравнения, наиболее часто хлорсеребряного (рис. 1). Внутренний раствор и внутренний электрод сравнения при измерении ЭДС ячейки остаются постоянными.
Зависимость потенциала электрода от активности потенциал-определяющего иона (электродная функция) можно выразить модификацией уравнения Нернста :
E = E0 ±Slg a,
где Е — измеряемая величина; S — тангенс угла наклона электродной функции.
Для разбавления растворов сильных электролитов, ионная сила которых поддерживается примерно постоянной добавлением фонового электролита, активность определяемой частицы можно заменить на ее концентрацию:
E = E0±S]gC.
Для однозарядного иона коэффициент наклона электродной функции равен 59,16 мВ на единицу измерения активности или концентрации определяемого иона в логарифмическом масштабе.
Минимальное количество вещества, которое еще можно определить с заданной достоверностью, называют пределом обнаружения метода. В потенциометрии предел обнаружения прежде всего зависит от растворимости материала мембраны в анализируемом растворе и обычно равен 10-6...10-7 M.
Селективность электродов, т. е. возможность определения того или иного иона в присутствии мешающих ионов, колеблется в очень широких пределах. Для количественной оценки влияния мешающих ионов на измерение концентрации 1-го иона вводят понятие коэффициента селективности Кij, численное значение которого определяется уравнением:
E = Ei0± |
RT |
ln (ai + ∑ ) Кij aj |
niF |
Помехи зависят от следующих причин. Прежде всего определению мешают другие потенциалопределяющие ионы. Кроме того, многие ионы или комплексообразующие реагенты образуют соединения с определяемым ионом, в результате изменяется его активность.
Важная характеристика ионоселективного электрода — время его отклика. В потенциометрии для характеристики быстродействия электрода используют понятие времени отклика, т. е. времени, необходимого для достижения постоянного значения потенциала электрода при перемещении его из одного раствора в другой, отличающийся по концентрации определяемого иона. Обычно для относительно концентрированных (10-4...10-2М) растворов время отклика не превышает 10—15 с, но для разбавленных (10-5М) может составлять несколько минут. В существенной степени время отклика зависит от типа электрода и обычно выше для жидкостных и пластифицированных электродов по сравнению с твердофазными.
Наиболее обоснованы названия электродов, для которых потенциалопределяющий и аналитически*' определяемый ионы совпадают. Например, если определяемый ион—катион меди (II), а мембрана электрода содержит сульфид меди (II) и потенциалопределяющий ион в этом случае — также катион меди указывать степень окисления нет необходимости, то такой электрод называют просто медьселективным электродом. Таким образом, в первой части названия — состав частицы, для измерения концентрации которой предназначен электрод. Слово «селективный» нельзя понимать буквально. В действительности значение коэффициентов селективности может быть больше единицы, и в этом случае электрод более селективен к мешающему иону, а не к определяемому. Поэтому можно применять термин «ионочувствительный электрод».
Аналитические характеристики электродов зависят от мембраны электрода и устройства его в целом. Поэтому при классификации электродов используют различные признаки. Прежде всего, электроды классифицируют по типу мембраны. Различают твердофазные, жидкостные и пластифицированные электроды, при этом имеют в виду электроды с твердой или жидкой мембранами и с жидкой мембраной, заключенной в матрицу из полимера, обычно поливинилхлорида. Конструкция электродов первого поколения обязательно содержала внутренний раствор сравнения и внутренний электрод сравнения.
Возможны и вполне работоспособные электроды, в которых ионочувствнтельная мембрана, непосредственно минуя раствор и электрод сравнения, контактирует с металлическим токоотводом. Такие электроды более технологичны при изготовлении и менее чувствительны к условиям эксплуатации. Общепринятого термина пока нет, часто такие электроды называют твердотельными. Иногда удобно объединять индикаторный электрод и электрод сравнения в единой конструкции. Такие объединенные электроды называют одностержневыми (рис. 2).
|
Рис. 2. Комбинированный стеклянный электрод к потенциометру ППМ-ОЗМ:
1 — насыщенный раствор КС!; 2 —асбестовая нить; 3 — хлорсеребряный электрод сравнения; 4 — соединительный кабель; 5 — защитный колпачок; 6 — порошок AgCl и KCI; 7 — отверстие с пробкой для доливки KCI; 8 — асбестовая нить; 9 — полиэтиленовая трубка; 10 — внутренний вспомогательный хлорсеребряный электрод в растворе HCI; 11 — шарик из электродного стекла
|

Твердофазные электроды. К электродам с твердыми мембранами относят прежде всего классический электрод для определения активности ионов водорода — стеклянный рН-электрод, изготовленный в начале нашего века. Ионочувствительной мембраной в этом случае служит тонкостенный шарик из специального стекла (содержащего, например, 72% оксида кремния, 8% оксида кальция и 20% оксида натрия), припаянный к обычной стеклянной трубке (рис. 3). Изображенные на рисунке 3 (а, б, в, г, д) электроды отличаются только формой и типом штекера для подсоединения к прибору. В шарик налит раствор хлористоводородной кислоты (0,1М), в который погружен хлор-серебряный электрод. Потенциал такого электрода зависит от
|
Рис. 3. Схемы электродов: а — селективного с внутренней системой сравнения; б — то же с твердым токоотводом; 1 — мембрана; 2 — металлический контакт; 3 — электрод сравнения; 4 — раствор сравнения; 5 — корпус электрода; в — стеклянного типа ЭСЛ-41Г-04; г — то же типа ЭСЛ-Ч1 Г-05; д — то же типа ЭСЛ-40-07: 1 — шарик из электродного стекла; 2 — раствор внутренний; 3 — внутренний электрод сравнения; 4 — корпус; 5 — выводной кабель; 6 — штекер; 7 — экран |

активности ионов водорода, или, точнее, ионов гидроксония. Стеклянные электроды обычно не рекомендуется использовать в щелочных растворах (рН 11), однако электроды из специального стекла пригодны и для работы в широком интервале концентрации ионов водорода (рН 0—14). Значение коэффициента селективности должно быть в этом случае меньше 10-14. В щелочной области наблюдается «отрицательная» погрешность (щелочная ошибка).
Определение и значение точной концентрации кислоты необходимо во многих случаях. Концентрация кислоты — важнейший параметр многих технологических процессов. Комфортные условия живых организмов также определяются кислотностью среды обитания и биологических жидкостей. Так, рН плазмы крови может колебаться всего лишь в интервале ±0,2 рН. К важнейшим глобальным загрязняющим веществам относят оксиды серы и азота. Повышенное содержание этих соединений в атмосфере приводит к увеличению кислотности дождевой воды — явлению, настолько распространенному, что выражение «кислотные дожди», к сожалению, стало общеизвестным.
Кроме чувствительного к ионам гидроксония (рН) электрода, разработаны стеклянные электроды, чувствительные к однозарядным катионам щелочных элементов, аммония, серебра, таллия и др.
В качестве чувствительного элемента для твердых нестеклянных мембран электродов используют вещества, которые обладают малой растворимостью и ионной проводимостью по катиону или аниону. К сожалению, этим требованиям отвечают немногие соединения, так как при комнатной температуре мало веществ обладает ионным характером проводимости. Такими свойствами обладают только фториды некоторых редкоземельных элементов и халькогениды очень небольшой группы элементов. В качестве мембран используются монокристаллы и мембраны, полученные прессованием или плавлением порошкообразных соединений и их смесей. Промышленность выпускает электроды, чувствительные к галогеннд-, цианид- и сульфид-анионам и к катионам серебра, меди, кадмия, свинца.
Один из важнейших электродов на основе твердых соединений с ионным характером проводимости — фторидный электрод, который обладает высокой селективностью по отношению ко всем ионам. В качестве мембраны в этом случае применяют монокристалл фторида лантана, в который для уменьшения сопротивления добавляют небольшие количества фторида европия. Высокая селективность фторидселективного электрода основана из том, что монокристалл обладает анионным характером проводимости.
Фторидселективный электрод работает в широком диапазоне активности анионов (от 10-7 до 1 М). Селективность мембраны настолько велика, что даже 1000-кратный избыток ионов галогена, а также нитрат-, фосфат- и гидрокарбонат-ионов не влияет на работу электрода.
Потенциал электрода зависит от активности или концентрации потенциалопределяющего иона, а не общей концентрации всех компонентов раствора, содержащих в составе этот ион. При использовании фторидселективных электродов необходимо, в частности, учитывать, что при снижении рН раствора идут реакции образования молекул фтористоводородной кислоты. В соответствии с этим активность аниона уменьшается, а потенциал электрода становится более положительным.
Распространен электрод из сульфида серебра. Такой электрод применяют для определения концентрации, как серебра, так и серы. Чрезвычайно малая растворимость, хорошая устойчивость по отношению к окислителям и восстановителям, достаточно высокая проводимость, а также простота получения методом прессования или плавления делают сульфид серебра идеальным материалом для изготовления электродов.
Учитывая очень низкое произведение растворимости сульфида серебра (ПР = 6-10-50), следовало бы ожидать очень большого рабочего диапазона для этого электрода. В действительности этот диапазон составляет 10-7...l М. С помощью буферных растворов можно существенно расширить интервал линейной зависимости градуировочного графика.
Газовый электрод широко применяют для определения концентрации аммиака в воде и увлажненном воздухе. Электрод позволяет также определять ионы аммония после перевода их в аммиак добавлением раствора гидроксида натрия к анализируемому раствору. Существуют также электроды для определения диоксида серы, оксидов азота, сероводорода, фтористого водорода, цианистого водорода.
Электроды с гетерогенными мембранами. В ионоселективных электродах этого типа твердое соединение, обладающее ионообменными свойствами, закрепляют в полимерной матрице мембраны. Например, в С1--селективном электроде хлорид серебра распределяют в парафине или силиконовом каучуке. Электрод, чувствительный к хлорид-ионам, был одним из первых ионоселективных электродов, но сейчас его применяют мало.
Фермент-субстратный электрод позволяет определять концентрацию ферментных субстратов или самого фермента. Например, предложенный Гилболтом и Монталво электрод на основе уреазы, чувствительный к мочевине, обеспечивает быстрое определение концентрации мочевины в растворе. Это стеклянный электрод, чувствительный к катионам, покрытый гелем, содержащем фермент уреазу (рис. 92). Гель поддерживает нейлоновая сетка («нейлоновый чулок»). При погружении электрода в раствор образца, содержащего мочевину, мочевина диффундирует в гель и происходит катализируемая уреазой реакция
С О (NH2)2+2Н2О + Н+ уреаз буфер, РН 7 2NН4+ + НС03-
После достижения равновесия в системе, на что требуется 30... 60 с, измеряют потенциал электрода. Электрод реагирует на образующийся NH4+; при постоянной концентрации уреазы и других стандартных условиях потенциал — линейная функция логарифма концентрации мочевины в растворе образца.
Разработаны и другие фермент-субстратные электроды. Например, для измерения концентрации аминокислот стеклянный электрод, чувствительный к катионам, покрывают слоем геля, содержащего оксидазу L-аминокислоты (L-AOO). Ионы аммония образуются по следующей реакции:
2 RCH+(NH3)COO- + O2 2RCOCOО- +2NH4+
Для разрушения образующейся при окислении аминокислот перекиси водорода добавляют еще один фермент — каталазу.
Существуют также аммиачные электроды. В них растворенный аммиак из образца диффундирует в электрод через проницаемую для газов мембрану. Равновесие между уровнями концентрации аммиака в образце и во внутреннем растворе устанавливается в течение одной минуты. Аммиак реагирует с внутренним раствором, образуя гидроксид-ионы, которые определяются чувствительным внутренним элементом:
N
 H3
+ H2O
NH4+
+ OH-.
H3
+ H2O
NH4+
+ OH-.
Зависимость электродного потенциала от логарифма концентрации аммиака линейна в интервале концентраций аммиака 10-1... 10-6 моль/л. Посторонние ионы, присутствующие в растворе образца, не мешают определению, исключение составляют лишь летучие амины.
Измерение потенциала.
Принципиальная схема устройства, применяемого для измерения потенциала — потенциометра, показана на рис. 4. Такие приборы также называют рН-метрами или ионометрами, так как они предназначены для измерения потенциалов ячеек, содержащих рН-чувствительный стеклянный электрод с высоким сопротивлением или рХ-селективные электроды аналогичных характеристик. Шкала этих приборов градуирована как в милливольтах, так и в единицах рН (рХ). Такие приборы удобны при измерении потенциалов ячеек с низким и высоким сопротивлением.
Измерение потенциала одного электрода невозможно, поэтому определяют ЭДС гальванического элемента, в котором, кроме индикаторного (ионоселективного) электрода, используют электроды сравнения с постоянным потенциалом. Наиболее удобны электроды второго род, где металл, покрытый слоем малорастворимой соли этого металла и находящийся в растворе, насыщенном этой солью и содержащем другую легко растворимую соль с тем же анионом).
|
Рис.4. Принципиальная схема измерения потенциала электролитических ячеек на рН-метре-милливольтметре: 1 — рН-метр-милливольтметр; 2 —чувствительный элемент; 3 — измерительный электрод; 4 — внутренний контактный электрод; 5 -внутренний электродный раствор сравнения; 5 — хлорсеребряный электрод сравнения; 7 — серебряная проволока; 8 — кристаллы AgCI; 9 - насыщенный раствор KCI; 10 — асбестовый фитиль; 11 — исследуемый раствор; 12 — контакт электролитического ключа |

В основе работы ионоселективных (мембранных электродов) лежат ионообменные реакции, протекающие на границах мембран с растворами электролитов, т. е. в электродных реакциях электроны участия не принимают. Действий мембранных электродов заключается в следующем. Мембрана, служащая основой электрохимической ячейки, селективна по отношению к данному иону. Она разделяет два раствора с различной активностью этого иона.
Теория стеклянного электрода была разработана Б. П. Никольским и основана на представлении о существовании обмена ионами между стеклом и раствором. Таким образом, электродная реакция заключается в обмене ионами Н+ между двумя фазами— стеклом и раствором.
В качестве внутреннего электрода сравнения в стеклянном электроде используют хлорсеребряный электрод. Наиболее часто стеклянный электрод изготавливают в виде стеклянной трубки с напаянной на конце мембраной в виде шарика (см. рис. 4, 5). Шарик заполняют раствором НС1 с определенной концентрацией.
На границе хлорсеребряного электрода и внутреннего раствора НС1 возникает совершенно определенный скачок потенциала. Последний возникает и на границе стеклянной мембраны с внутренним раствором. В ходе измерения рН активная концентрация внутреннего раствора, а значит, и сумма внутренних скачков потенциала остаются постоянными.
Графически зависимость ЭДС гальванического элемента со стеклянным и вспомогательными электродами от рН в области водородной электродной функции стеклянного электрода представляет собой прямую линию (рис. 5, а).
|
Рис. 5. Зависимость потенциала (Е) стеклянного электрода от рН среды (а) и от температуры (б): 1 — зона кислотных погрешностей; 2 — рабочая зона; 3 — щелочные погрешности; 4 — изотерма при 100 ○С; 5 — изотерма при 50 °С; 6 — изотерма при 0 ○С; 7 — изопотенциальная точка. |
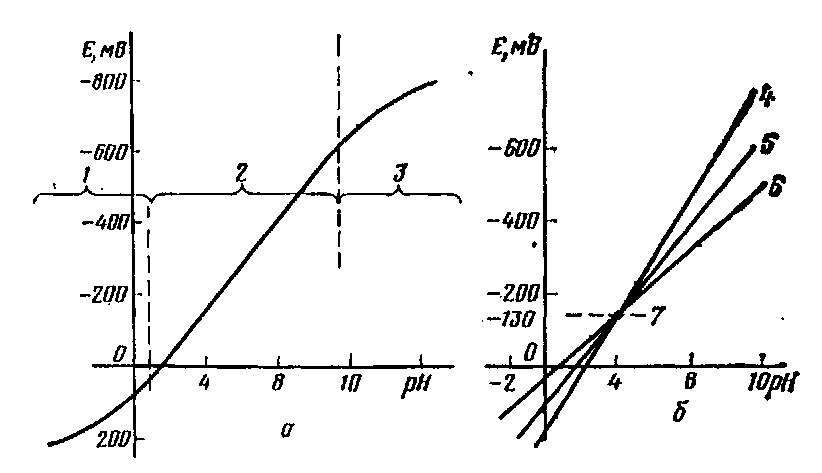
В очень кислых растворах могут наблюдаться «кислотные ошибки», в очень щелочных— «щелочные ошибки», т. е. кривая отклоняется от линейного хода. Положение этих отклонений зависит от сорта стекла и состава ионов. Уравнение этой прямой
E = K+b · lg aH+ = K—b pH,
где K—(R·T·2,303)/F, Е — потенциал электрода; R — универсальная газовая постоянная; Т — температура, в градусах абсолютной шкалы Кельвина; F — постоянная Фарадея; 2,303—модуль перехода от натуральных логарифмов к десятичным; а, b — коэффициенты.
Из уравнения видно, что прямая отсекает на оси ЭДС (ордината), проходящей через рН 0, отрезок, равный К мВ, и идет под углом, тангенс которого равен b. Положение прямой в координатах Е — рН для каждого электрода устанавливают калибровкой по стандартным растворам с известным и устойчивым значением рН — буферным растворам. Для калибровки достаточно двух растворов, но лучше, чтобы их было не менее трех. Установив положение прямой, можно далее по графику или по откалиброванной шкале прибора узнать рН любого раствора, если опустить в него откалиброванный стеклянный электрод с тем же вспомогательным электродом, что и при калибровке, и измерить ЭДС.
Как видно из уравнения и рис. 5, б, зависимость потенциала электрода от температуры есть величина линейная. Однако существует такое значение рН (изопотенциальная точка электрода), при котором потенциал электрода не зависит от температуры. При настройке прибора учитывают эту величину, а также величину и знак температурной поправки при значениях рН, отличных от величины изопотенциальной точки. Перед измерением рН прибор настраивают по буферным растворам той же температуры, что и контролируемый раствор.
Основные типы буферных растворов, используемых для стандартизации рН-метра: 0,05 М раствор тетраоксалата калия (рН 1,69 при температуре 25°С); 0,025 М раствор однозамещенного фосфата калия и 0,025 М раствор двузамещенного фосфата натрия (рН=6,86 при температуре 25°С); 0,01 М раствор тетра-бората натрия (рН = 9,18 при температуре 25°С). В диапазоне измерения рН 0... 11 абсолютная погрешность измерения составляет ±0,01 рН (±0,58 мВ); в диапазоне 0... 14—±0,1 рН (±5,8 мВ).
Для анализов широко используют различные рН-метры, например рН-121, иономер ЭВ-74, иономер переносной И-102 и др. Выпускают вольтметры прямого отсчета, снабженные электронными усилителями для получения сигнала, который можно регистрировать с помощью цифрового счетчика (И-120). Эти приборы пригодны для работы с электролитическими ячейками, обладающими низким и высоким сопротивлением; рН-метры могут использоваться в качестве нуль-индикатора и высокоомного милливольтметра. В комплект приборов входят и ячейки для термостатирования, дополнительные электролитические ключи для замены КС1 при определении образцов, в которых КС1 мешает.
Применение ионоселективных электродов для определения концентрации ионов в водных растворах.
Методы определения в водных растворах макро- и микроконцентраций ионных компонентов и их соединений с помощью ионоселективных электродов (ИСЭ) отличаются и техникой применения последних, и аналитическими приемами. При анализе водных растворов с широкой вариацией химического состава неизбежна разработка новых и совершенствование известных методик в соответствии с аналитическими требованиями и конкретными условиями применения ИСЭ.
Например, определение содержания одного и того же ионного компонента в водах различного происхождения (в поверхностных и загрязненных промышленных водах) может существенно различаться. Это зависит от различия в содержании определяемых ионов, соотношения концентраций определяемых и мешающих ионов, рН среды и др., что обусловливает необходимость изменения техники аналитического применения ИСЭ.
Основные преимущества прямой потенциометрии — достаточно высокая надежность, универсальность и быстрота определений.
Она обеспечивает:
— определение активности (термодинамической концентрации) отдельного иона в многокомпонентных растворах;
— выполнение микроанализа в пробах объемом в десятые доли миллилитра;
— исследование кинетики реакций;
— выполнение определений не только в стационарных (лаборатории, станции контроля), но и в полевых условиях;
— в сочетании с другими методами анализа существенное увеличение эффективности определений;
— выполнение определений в мутных и окрашенных растворах и даже в вязких пастах;
— измерение in situ при контроле самых различных объектов и процессов (гидрохимические, экологические, медико-биологические исследования, технологические процессы в производстве и др.);
— непосредственную связь информативного параметра (ЭДС электродной системы) с активностью определяемого иона. Это позволяет довольно легко автоматизировать аналитические определения с помощью средств, аналогичных для других методов, основанных на обработке электрических сигналов.
Технические средства для потенциометрических измерений просты в изготовлении и эксплуатации. Материальные затраты на измерительную аппаратуру и химреактивны для исследований с помощью ИСЭ незначительны.
Подготовка проб. Практически невозможно дать подробные рекомендации, имеющие универсальный характер. Поэтому ниже приведены только наиболее важные принципы, которые необходимо соблюдать при подготовке проб.
Ионная сила растворов. При измерении ионной концентрации необходимо учитывать ионную силу растворов. Это обусловлено тем, что ИСЭ чувствительны к активности ионных компонентов, которая, в свою очередь, функционально связана с концентрацией ионов в растворе. Связь эту выражают коэффициентом активности, величина которого в основном зависит от ионной силы раствора.
Основной принцип определения концентрации ионных компонентов с помощью ИСЭ — поддержание постоянного значения ионной силы всех растворов, применяемых в аналитическом процессе. Для этого в анализируемый раствор вносят в определенном соотношении индифферентный электролит с достаточно большой ионной силой.
Химическая подготовка растворов. Так как ИСЭ реагирует на активность определяемого иона, который находится в диссоциированном состоянии, для определения «полной» концентрации нужно перевести определяемые ионы, связанные в комплексы или ионные пары, в свободное диссоциированное состояние. Это довольно сложная аналитическая задача, для решения которой необходимо предположительно знать состав анализируемого раствора.
При соответствующих условиях возможно также определение тех элементов, которые в растворе имеют различную степень окисления. Обеспечив в анализируемом растворе окислительные или восстановительные условия (методом введения определенных ингредиентов), в ходе реакций можно перевести ионы из одной степени окисления в другую.
Учитывая, что ИСЭ имеют ограниченную селективность по отношению к мешающим ионам, для повышения надежности определений принимают меры, снижающие влияние этих ионов на электродную функцию ИСЭ. В анализируемый раствор вводят реагенты, уменьшающие влияние мешающих ионов. Обычно либо осаждают мешающие ионы, либо вызывают образование малодиссоциированных комплексов, удерживающих эти ионы.
Значение рН растворов. При выполнении большинства определений с помощью ИСЭ необходимо поддерживать в анализируемом растворе оптимальное значение рН. В противном случае возможно проявление мешающих Эффектов первого порядка из-за чувствительности ИСЭ к ионам Н3О+ или ОН- (например, для стеклянных электродов, чувствительных к ионам Na+, K+, NH4+), а также мешающих эффектов второго порядка из-за уменьшения концентрации определяемых свободных ионов, которые реагируют с ионами H3O+, ОН-
Методы поддержания оптимального значения рН пробы разнообразны. Чаще всего для этого применяют буферные растворы, которые слабо диссоциируют в воде и поэтому не мешают определению (например, ацетатный буфер). Также применяют различные щелочи и кислоты (NaOH, КОН, NH4OH, HC1 и др.).
Индифферентные электролиты. На практике вышеперечисленные принципы подготовки анализируемых растворов стремятся осуществить путем совмещения всех функций в одном индифферентном электролите, который представляет собой буферную среду с высокой ионной силой, содержащую сильный комплексообразующий реагент для уменьшения влияния мешающих ионных компонентов.
К индифферентному электролиту предъявляют следующие требования:
этот электролит не должен содержать мешающих ионов и компонентов, вредно воздействующих на поверхность ИСЭ;
значение его рН должно быть в диапазоне оптимальных значений для определений;
буферность индифферентного электролита должна быть значительной, чтобы обеспечить постоянную активность водородных ионов после смешивания с анализируемым образцом.
Общие требования к комплексу технических средств и реактивам. Для измерений ЭДС электрохимических цепей можно использовать любой милливольтметр с высоким сопротивлением RBX≥ 1011Ом), (погрешность измерений которого не более 1 мВ.
Для перемешивания растворов, особенно в случае применения индифферентного электролита, необходимо применять магнитные мешалки с регулируемой скоростью вращения.
Определение градуировочной характеристики. Эмпирическое определение градуировочной характеристики электродной системы необходимо для снижения погрешности результата анализа, возникающей из-за ненадежности определения потенциала электрода сравнения, жидкостных диффузионных потенциалов, а также из-за влияния других мешающих факторов: температуры, изменения ионной силы раствора.
Из стандартного 0,1 М раствора методом разбавления дистиллированной водой готовят серию растворов, отличающихся 10-кратным разбавлением. Сливают в определенном соотношении (чаще всего 1:1) точно отмеренные объемы (10—50 л) стандартного раствора и индифферентного электролита и тщательно перемешивают магнитной мешалкой.
Электродную систему последовательно погружают во все приготовленные калибровочные растворы и измеряют ЭДС электродной системы в каждом растворе. Показания прибора считывают после установления равновесного потенциала ИСЭ. Время установления равновесного потенциала в первом приближении зависит от содержания определяемого ионного компонента в растворе и значения ионной силы. При содержании ионного компонента 10-3 М и ионной силе i ≈ 0,5 M время установления равновесного потенциала равно 10—30 с. При более низком содержании ионного компонента время увеличивается до нескольких минут.
Затем строят калибровочный график, который обычно представляет собой прямую линию в области высоких концентраций и отображает линейную зависимость ЭДС электродной системы от логарифма концентрации определяемого иона. При низких концентрациях определяемого иона наблюдается нелинейный характер зависимости ЭДС электродной системы от содержания этого иона. Для точного построения калибровочного графика необходимо произвести определения в промежуточных точках. Например, 2·10-6, 4·10-6, 6·10-6 М стандартного раствора (рис. 6).
|
Рис. 6. Графическое выражение зависимости величины потенциала (Е) натрий-селективного электрода от pNa; pNax —неизвестная концентрация; pNai и pNai+1 — известные концентрации
|
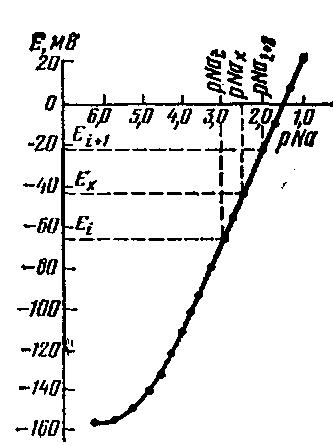
Если ориентировочно известна концентрация определяемого иона в анализируемом растворе, то не нужно строить градуиро-вочный график во всем диапазоне электродной функции ИСЭ. В таком случае достаточно приготовить серию стандартных растворов таким образом, чтобы концентрации ионных компонентов в стандартных растворах охватывали весь диапазон ожидаемых концентраций в анализируемых растворах.
При выполнении операций определения градуировочной характеристики рекомендуется соблюдать следующие условия:
температуры стандартных и анализируемых растворов должны быть равными;
ионная сила стандартных и анализируемых растворов должна быть идентичной;
градуировочную характеристику желательно определить в стандартных растворах, содержащих, кроме исследуемого иона, те же ионные компоненты, что и анализируемый раствор;
определение градуировочной характеристики необходимо начинать с растворов, имеющих более низкую концентрацию, переходя к стандартным растворам с более высокой концентрацией.
В результате определения градуировочной характеристики находят частные значения функциональной зависимости ЭДС электрохимической цепи от содержания анализируемого ионного компонента в растворе. Однако в отличие от «классических» методов анализа (гравиметрия, титриметрия), для которых характерны постоянство и неизменность во времени стехиометрических коэффициентов, в потенциометрическом анализе с применением ИСЭ определение градуировочной характеристики должно быть непосредственно связано во времени с исследованием содержания ионного компонента в анализируемом растворе (из-за нестабильности градуировочной характеристики). Необходимо периодически контролировать градуировочную характеристику. Рекомендуется осуществлять контроль после 10—20 определений.
ДРУГИЕ ВИДЫ ФИЗИКО-ХИМИЧЕСКИХ МЕТОДОВ АНАЛИЗА