

1. Электрохимические и полярографические методы
Теоретические основы и классификация
Электрохимия, изучающая взаимосвязь между электрической энергией и химическими реакциями, находит в физико-химических методах анализа разнообразное применение. Электрохимические методы анализа основаны на использовании электрохимических процессов, происходящих в электролитической ячейке (гальваническом элементе). Электролитическая ячейка представляет собой электрохимическую систему, состоящую из электродов и электролитов, контактирующих между собой.
Различают два основных типа электрохимических ячеек. К первому типу относится гальванический элемент, или ячейка с напряжением.
Химические реакции, протекающие внутри гальванического элемента, вызывают электрический ток. В схеме, изображающей элемент (рис. 1, а), электрод с более отрицательным электродным потенциалом записывают слева, с более положительным — справа:
Граница твердой и жидкой фаз показана одной вертикальной линией. Две линии указывают на границу между двумя жидкими фазами (жидкостное соединение) или на наличие солевого мостика. Вертикальные линии указывают на границы возникновения потенциалов в электрохимической цепи.
|
Рис. 1. Схемы гальванического элемента (а) и электролитической ячейки (б): 1 - цинковый электрод; 2 - гальванометр; 3 — медный электрод; 4 - солевой мостик; 5 — батарея; 6 — платиновый электрод |
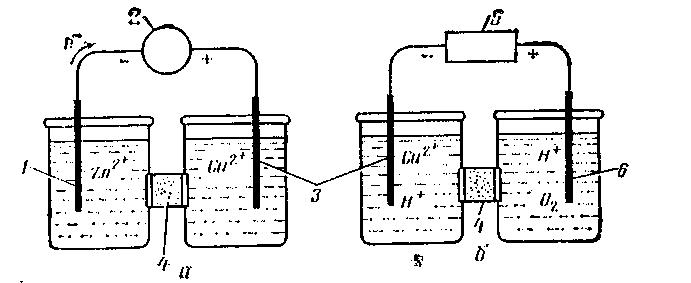
Другой тип ячейки — электролитическая. Химическая реакция идет в ней при наложении внешнего напряжения, которое противодействует току (если он есть) гальванического элемента (рис.1,б).
В электролитической ячейке медный электрод имеет отрицательный заряд, а платиновый — положительный:
C u2+
+ 2ē
Cu,
Е0
= 0,345 В
u2+
+ 2ē
Cu,
Е0
= 0,345 В
Н 2о
½ О2+
2Н+
+ 2ē, Е0
= 1,229 В
2о
½ О2+
2Н+
+ 2ē, Е0
= 1,229 В
Под действием приложенного напряжения электроны перемещаются от отрицательно заряженного электрода к положительному. Это приводит к обратимости электродных реакций но сравнению с реакциями в гальванической цепи. Общая реакция в электролитической ячейке — это сумма реакций, протекающих на двух электродах:
C
 u2+
+ Н20
Cu
(тв.) + ½ О2
+ 2Н+
u2+
+ Н20
Cu
(тв.) + ½ О2
+ 2Н+
Таким образом, в электрохимической ячейке отдельные электродные реакции и суммарная реакция ячейки имеют направление, противоположное самопроизвольной реакции гальванического элемента. Минимальный потенциал, необходимый для того, чтобы вызвать электролиз, должен быть выше потенциала гальванического элемента и иметь противоположное направление.
В состав электролитической ячейки могут входить два или три электрода, один из которых — индикаторный или рабочий, второй — электрод сравнения, третий— вспомогательный. Электрод, действующий на датчик, реагируя на состав раствора (не оказывая влияния на состав раствора во время измерения), — индикаторный. Если под действием тока, проходящего через ячейку, наблюдается значительное изменение состава раствора, то электрод — рабочий. Электрод сравнения служит для создания измерительной цепи и поддержания постоянного значения потенциала индикаторного (рабочего) электрода.
Используемый в трехэлектродной ячейке вспомогательный электрод (противоэлектрод) вместе с рабочим электродом включен в цепь, через которую проходит электрический ток. В состав электролитической ячейки могут входить два идентичных электрода сравнения.
В некоторых случаях на электродах электрохимической ячейки электрохимические реакции не проходят, и электроды служат для контакта между Проводником первого и второю рода, например, при определении электропроводности. Электрохимические методы анализа основаны на использовании зависимости электрических параметров электрохимических цепей от концентрации, природы и структуры вещества, участвующего в электродной (электрохимической) реакции или в электрохимическом процессе переноса зарядов между электродами. Электрохимические методы анализа можно подразделить на: методы без протекания электродной реакции, в которых строение двойного электрического слоя в расчет не принимают (кондуктометрия при низких и высоких частотах); методы, основанные на электродных реакциях в отсутствие тока (потенциометрия) или под током (вольтамперометрия, кулонометрия, электрогравиметрия).
Электрохимические методы в зависимости от типа электрохимической ячейки можно разделить на несколько групп. К первой группе относят потенциометрию и потенциометрическое титрование, где используют ячейки первого типа (гальванический элемент). Такими методами ведут прямое определение концентрации веществ в агрохимических исследованиях с помощью селективных электродов. Ко второй группе относят электрохимические методы исследования с применением электролитической ячейки (электрогравиметрия, кулонометрия, вольтамперометрия и др.). В третью группу можно объединить такие методы, в которых электрохимическая ячейка работает по двум направлениям — и как электролизер, и как гальванический элемент (например, хронопотенциометрия). В четвертую группу входят кондуктометрия, низко- и высокочастотное кондуктометрическое титрование.
Во всех электрохимических методах используют электрические параметры (сила тока, напряжение, сопротивление) как аналитический сигнал. Эти параметры не зависят от цветности растворов, агрегатного состояния. Электрические параметры можно измерить непосредственно в биологических объектах с достаточной точностью. Данные методы применяют либо для прямых измерений, основанных на зависимости «аналитический сигнал—состав», либо для индикации конечной точки титрования в титриметрии. Электрохимические методы анализа позволяют определять концентрацию вещества в широком интервале (1...10-9 моль/л) с достаточной точностью и воспроизводимостью. Эти методы можно автоматизировать для непрерывного контроля за процессами, проходящими в почве, растениях. В данной теме мы рассмотрим кулонометрию, полярографию и инверсионно-хронопотенциометрию.
Кулонометрические методы анализа
Кулонометрия основана на измерении количества электричества, израсходованного на электролиз определяемого вещества при постоянном потенциале. Название «кулонометрия» связано с измерением в эксперименте количества электричества, израсходованного на восстановление или окисление исследуемого вещества, в кулонах. Данный метод позволяет определять малые количества вещества, не поддающиеся определению обычными методами (весовым и объемным).
Прямую кулонометрию осуществляют при постоянном потенциале или силе тока. При этом определяемое вещество непосредственно окисляется или восстанавливается на электроде.
Косвенный метод кулонометрии — кулонометрическое титрование, при котором точка эквивалентности соответствует моменту, когда сила тока электролиза достигает величины «фонового» тока. При кулонометрическом титровании не прибавляют стандартный раствор титранта, как это делают при других способах титрования. Реагент образуется за счет внутренней и внешней электролитической генерации. Например, электрогенерированными окислителями могут быть ионы железа (III), церия (VI), хрома (VI), а также элементные: йод, бром и др. В качестве электрогенерированных восстановителей применяют (Ионы олова (II), хрома (II), железа (II), йода (I) и др.
В методах, основанных на реакциях кислотно-основного титрования, в кулонометрии водных растворов применяют электрогенерированные в процессе электролиза воды ионы водорода, образующиеся в прианодном пространстве, и ионы гидроксила, накапливающиеся в прикатодном пространстве в результате следующих реакций электрохимического восстановления-окисления воды:
н а
катоде: 2Н2О
+ 2ē - Н2+2ОН-
а
катоде: 2Н2О
+ 2ē - Н2+2ОН-
на аноде: Н2О - 2ē ½ О2 + 2Н+
Согласно закону Фарадея, количество вещества, превращаемого при электролизе, прямо пропорционально силе тока и продолжительности его прохождения. Поэтому в кулонометрическом титровании вместо объема и массы обычно измеряют время и величину тока, расходуемого на электрохимическую реакцию, сопровождающуюся образованием электрогенерируемых веществ.
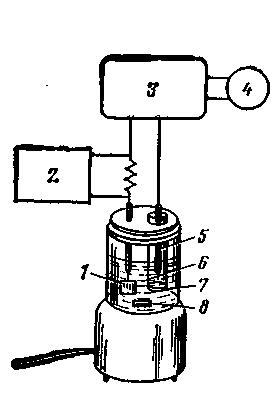
Кулонометрический анализ выполняют либо при постоянном контролируемом потенциале рабочего электрода, обеспечивающем 100 %-ю эффективность электрического тока (потенциостатическая кулонометрия, или потенциостатический метод), либо при постоянной силе тока (метод ам-перостатического кулонометрического титрования). Схема установки для кулонометрического титрования представлена на рисунке 2
Кулонометрическое титрование отличается высокой точностью и большой чувствительностью, что позволяет применять этот метод для определения микропримесей и малых количеств определяемого
вещества. Метод характеризуется экспрессностью, не требует высокой квалификации экспериментатора и отличается известной селективностью (особенно потенциостатическая кулонометрия). При использовании этого метода отпадает необходимость в стандартизации растворов и приготовлении калибровочных графиков по образцам с известным содержанием определяемого вещества.
|
Рис. 2. Кулонометрнческая установка: 1 — измерительный электрод; 2 - потенциометр; 3 - источник постоянного тока; 4- электронные часы; 5 - вспомогательный электрод; 6 — электролит; 7 - диск из спеченного стекла; 8 - мешалка
|
Полярография
Полярография основана на измерении силы тока, меняющейся в зависимости от приложенного напряжения в процессе электролиза, в условиях, когда один электрод (катод) имеет очень малую поверхность (поляризующийся), а другой (анод) - большую (неполяризующийся). Поляризующимся катодом могут служить капли ртути, вытекающие из тонкого отверстия капиллярной трубки, а также платиновый (вращающийся), графитовый и другие электроды. Неполяризующимся анодом служат стандартные электроды сравнения с большой поверхностью. Силу тока, при которой происходит полный разряд всех ионов анализируемого вещества, поступающих в приэлектродное пространство вследствие диффузии, называют предельным диффузионным током. Величина этого тока пропорциональна исходной концентрации определяемого вещества (ионов) в растворе. Основные принципы. Данный метод анализа основан на явлении концентрационной поляризации при протекании тока через раствор и получении так называемых вольтамперных кривых. Эти кривые получают при электролизе электровосстанавливающегося или электроокисляющегося вещества в электролитической ячейке специальной конструкции (рис. 3 , а). Один из электродов должен быть с малой поверхностью (обычно капельный ртутный электрод), другой — с большой. При пропускании через раствор постоянного тока происходит основное изменение концентрации у электрода с малой поверхностью. Это объясняется большой силой тока на единицу поверхности малого электрода, т. е. высокой плотностью тока на микроэлектроде.
Поверхность электрода сравнения (неполяризующегося электрода) должна быть неизмеримо больше поверхности индикаторного электрода. На большой поверхности электрода сравнения плотность тока довольно мала, поэтому вблизи этого электрода изменение концентрации очень невелико и не влияет на кривую зависимости силы тока от приложенного напряжения.
|
Рис. 3. Полярографичеекий анализ: а — схема строения полярографической ячейки; б—электрическая цепь для полярографического анализа: 1 — исследуемый раствор; 2— инертный газ (азот); 3 — ртуть; 4 — капельный ртутный электрод; 5 — солевой мостик; 6 — хлорид калия; 7 — каломель; 8 —батарея аккумуляторов на 4...6 В; 9 — реохорд; 10 — подвижный контакт; 11 — гальванометр; 12 — полярографнческая ячейка |
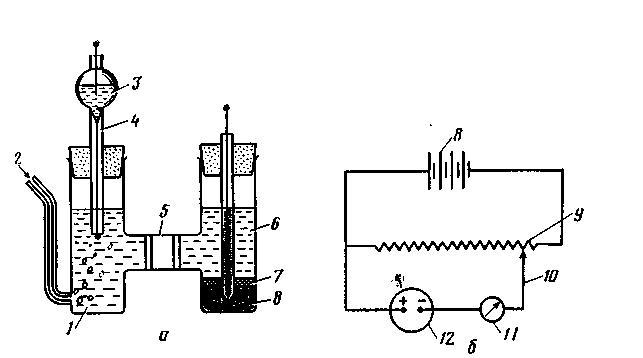
На электроды подают постепенно увеличивающееся напряжение с помощью внешнего источника тока (рис. 3, б). Если на ось абсцисс наносить величину приложенной ЭДС (Е), а на ось ординат — соответствующую силу тока (I), то получается S-образная кривая, которую называют полярографической волной (рис. 4,а).
По характеру полярографической волны можно определить как состав, так и концентрацию электровосстанавливающегося или электроокисляющегося вещества (деполяризатора). Таким образом, полярографический метод можно применять как для качественного, так и для количественного анализа.
|
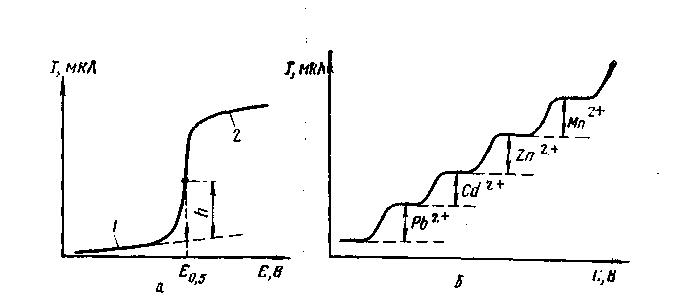
Рис. 4 Полярографическая кривая (а) и поляролграмма (б) раствора, содержащего катионы Pb2+, Cd2+, Zn24, Mn24: 1 - остаточный ток; 2 - диффузный ток, h — высота полярографической волны; Е0,5 - потенциал полуволны; I — сила тока; E —потенциал ячейки |
Для повышения электрической проводимости к исследуемому раствору добавляют раствор какого-либо электролита, содержащего катион с высоким потенциалом восстановления (обычно раствор соли щелочного или щелочноземельного металла). При этом перенос тока происходит только за счет движения ионов этого электролита. Определяемые ионы, поскольку концентрация их намного меньше ионов электролита, участвуют в этом переносе незначительно, появление их в приэлектродном пространстве обусловлено только процессом диффузии из более отдаленных частей раствора. Растворы электролитов, с помощью которых устраняют влияние электрического поля, называют фоновыми растворами, или фоном.
При наличии фона в исследуемом растворе полярографическая кривая показывает, что до тех пор, пока приложенное напряжение не достигло некоторой определенной величины, сила тока остается постоянной, близкой к нулю (остаточный ток), но как только напряжение превысит эту величину, сила тока очень быстро возрастает с увеличением напряжения, и кривая круто поднимается вверх. Однако возрастание силы тока вскоре прекращается, и кривая переходит в прямую, параллельную оси абсцисс (предельный диффузионный ток). Если из средней точки этого участка кривой опустить перпендикуляр на ось абсцисс, то получают значение напряжения, называемое потенциалом полуволны. Эта величина не зависит от концентрации и однозначно характеризует природу восстанавливаемых ионов. Таким образом, по величине потенциала полуволны можно определить присутствующий в растворе ион.
Если в растворе присутствует не один ион, способный восстанавливаться на катоде, то при дальнейшем повышении напряжения после достижения предельного диффузионного тока первого катиона через некоторый интервал времени будет достигнут потенциал, при котором начинают восстанавливаться катионы другого металла. Следовательно, вольтамперная кривая после горизонтального участка начнет снова круто подниматься вверх, и за первой полярографической волной последует вторая, за ней третья, если присутствует третий катион, и т. д. Если потенциалы восстановления этих ионов различаются на достаточно большую величину (не менее чем на 0,2 ), то ноны могут быть открыты качественно и определены количественно по полученной полярограмме. На рисунке 4, б приведена полярограмма для раствора, содержащего катионы РЬ2+, Cd2+, Zn2+, Mn2*.
Количественное определение основано на измерении высоты полярографической волны, т. е. величины предельного диффузионного тока. По мере увеличения напряжения скорость восстановления ионов определяемого металла на катоде непрерывно возрастает и непосредственно прилегающий к катоду слой раствора все более обедняется этими ионами. В конечном итоге система достигнет такого состояния, когда количество ионов, разряжающихся на катоде в единицу времени, равно количеству ионов, которые подходят к катоду в результате диффузии из более отдаленных частей раствора. Начиная с этого момента больше не наблюдается дальнейшего увеличения силы тока с возрастанием напряжения. Получаемый предельный ток, обусловленный скоростью диффузии, называется также диффузионным.
Скорость диффузии пропорциональна разности концентраций ионов, в случае предельного тока она равна нулю. Высота полярографической волны (h) прямо пропорциональна концентрации восстанавливающегося на катоде (т. е. определяемого) иона в растворе.
Инверсионно-хронопотенциометрический метод
Классические электрохимические методы анализа, в основе которых лежит стационарная (поляризационная кривая, имеют предел обнаружения вещества в очень малых количества (микрограммы).
Однако такая чувствительность недостаточна для решения ряда современных экологических задач, определения состояния микроэлементов в почве, в объектах сельскохозяйственного производства, пищевом сырье, сточных водах и.т. д. Снизить пределы обнаружения на несколько порядков можно, предварительно концентрируя разбавленный раствор образца. Для этого применимы некоторые методы, например хроматография или жидкостная экстракция,
К недостаткам таких методик относятся продолжительность анализа и трудоемкость, возможность потери части определяемого вещества при концентрировании (выпаривании, экстракции и др.) и введения загрязнений и дополнительных компонентов в анализируемую систему. Удобнее осуществлять предварительное концентрирование без внесения дополнительных веществ, т. е. прямо в системе, в которой будут выполнять измерение. На таком принципе основано концентрирование макроколичеств веществ при электрохимических инверсионных методах анализа.
Сущность метода. Метод инверсионной хронопотенциометрии заключается в предварительном электрохимическом концентрировании исследуемого элемента на рабочем электроде при заданном потенциале и поступлении этого раствора из перемешиваемого раствора ячейки на калиброванное внешнее сопротивление. При этом накопленный элемент переходит в раствор. Так как на электроде исследуемое вещество находится в концентрации, во много раз превышающей концентрацию его и первоначальном растворе, чувствительность определения возрастает.
В основе метода лежит определение продолжительности растворения накопленного элемента (переходное время), которое при постоянных условиях концентрирования и растворения пропорционально концентрации элемента в растворе.
Величина переходного времени определяется уравнением
τ = KSCtR,
где τ - -переходное время; К—электрохимическая константа для данного элемента; S — площадь рабочего электрода; t — время концентрирования элемента; С — концентрация элемента в растворе; R — заданное внешнее нагрузочное сопротивление.
Рабочая схема прибора (рис. 5) включает две самостоятельные электрические цепи а и б. В цепи а размещен источник постоянного напряжения 12 (аккумулятор), от которого с помощью потенциометра 11 отбирают необходимое напряжение для электролитического концентрирования определяемого элемента. Напряжение контролируют вольтметром 10. Исследуемый раствор помещают в ячейку 9, в которой размещены рабочий индикаторный электрод 7 и электрод сравнения 5, например, насыщенный каломельный электрод.
|
Рис. 5. Принципиальная схема инверсионно-хронапотеициометрического метода: а — цепь с источником постоянного тока; б — цепь с нагрузочным сопротивлением: 1, 3 — клеммы тумблера; 2 — тумблер; 4 —дифференцирующий конденсатор; 5 — нагрузочное сопротивление; 6 — низкочастотный осциллограф; 7 — индикаторный электрод; 8 — электрод сравнения; 9 —ячейка; 10 — вольтметр; 11—потенциометр: 12 ~- источник постоянного напряжения |
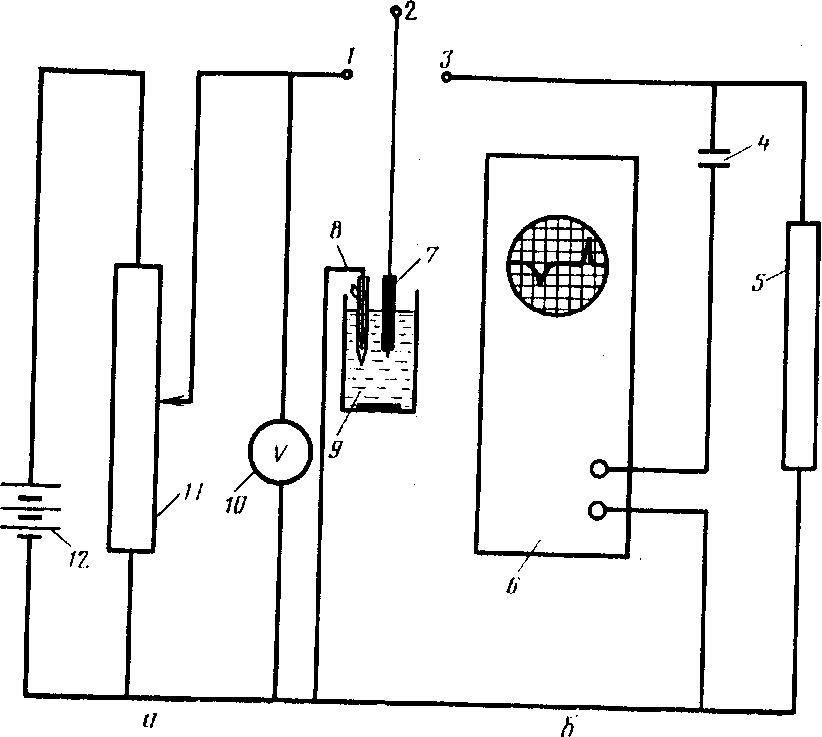

Метод инверсионной хронопотенциометрии в основном применяют для определения ионов металлов, поэтому, когда тумблер 2 находится в положении 1, происходит электролиз, и определяемый элемент осаждается на рабочем электроде:
M e0
– ne-
Me0
e0
– ne-
Me0
Цепь б содержит нагрузочное сопротивление 5, регулирующее скорость растворения, и регистрирующий прибор — низкочастотный осциллограф 6. Осциллограф такого типа предназначен для регистрации низкочастотных электрических сигналов, начиная ,с 0,1 Гц и выше, и позволяет исследовать импульсные процессы постоянного тока длительностью от долей миллисекунды до десятков секунд.
За счет высокого входного сопротивления — 0,5 мОм — осциллограф не влияет на режим исследуемых процессов. Чувствительность усилителя вертикального отклонения, регистрирующего величину потенциала электрода, устанавливают на максимальную величину — 5 мВ/мм, при необходимости ее можно регулировать. Точную калибровку вертикальной развертки выполняют с помощью внешнего источника напряжения. Исследуемый сигнал подается на вход прибора через коаксиальное гнездо и дифференцируется с помощью конденсатора 4,
По истечении времени электролитического концентрирования тумблер из положения 1 перемещают в положение 2, и происходит электрохимическое растворение выделенного на электроде металла
М е° - ne- Меn+
Потенциал рабочего электрода в процессе растворения зависит от индивидуальных электрохимических свойств элемента. Как только растворение завершится, потенциал рабочего электрода возвращается к своему первоначальному значению. Изменение потенциала рабочего электрода во времени отражается на экране осциллографа в виде дифференциальной кривой растворения dE/dτ—τ. Переходное время регистрируют с точностью до 0,1 с секундомером или электрохронометром, включенным в цепь растворения.
При электроосаждении на рабочем электроде атомов нескольких металлов электрохимическое растворение их осуществляется индивидуально, начиная с наиболее электроотрицательного. После того как растворится самый электроотрицательный металл, потенциал электрода падает до значения следующего за ним более электроположительного металла и так далее (рис. 6).
|
Puc. 6. Осциллограммы производных кривых растворения: а — РЬ; б - Cd, Pb, Си; а ~-Мп. Zn, Cd, Pb, Си; условия накопления для вариантов: о — фон — t Н. HCI; tэл = 1 мин; Еэл= - 0,8 В; R = 70 кОм: СрЬ+2=4·-10-6 г-ион/л; б — фон — 1 н. HCI: tэл = 1 мин; Еэл = - 1 В; R = 70 кОм; С Cd2+ = 1,5 ·10-5 г-ион/л; C Рb2+= 6-10-6 г-нон/л; C Си2+= 1.8-10-5 г-ион/л; в - фон 1 н. KCI; tэл =30 с; Еэл = —1.65 В; R =70 кОм; C Mn2+= = 1-10-4 г-ион/л; C Zn2+= 2-10-5 г-ион/л; С Cd2+=1,5-10-5 г-ион/л; C Рb2+= = = 2-10-6 г-ион/л; C Си2+= 1,8-10-5 г-ион/л: tэл — время электролиза; Еэл — потенциал; τMe — переходное время растворения; R — величина нагрузочного сопротивления;h -высота пика; С — концентрация соответствующего элемента |
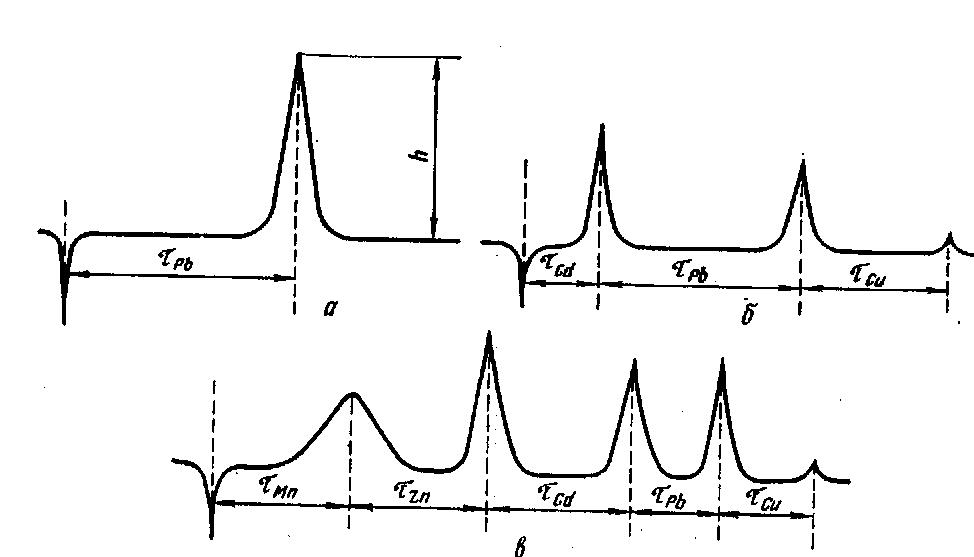
Время перемещения луча на экране осциллографа между вершинами смежных пиков — это переходное время электрохимического растворения соответствующего металла. После растворения всех элементов электроды замыкают для полной их регенерации и подготовки к следующему измерительному циклу.
Полный цикл электрохимической инверсии представлен на диаграмме (рис. 7).
|
Рис. 7. Диаграмма смены режимов поляризации электрода: Еэл — потенциал электролиза; τэл —время электролиза; τMe -•-переходное время электрохимического растворения металла; τр — время регенерации электрода при коротком замыкании; τц- общая длительность цикла (вертикальными пунктирными линиями обозначено время, в течение которого раствор не перемешивается)
|
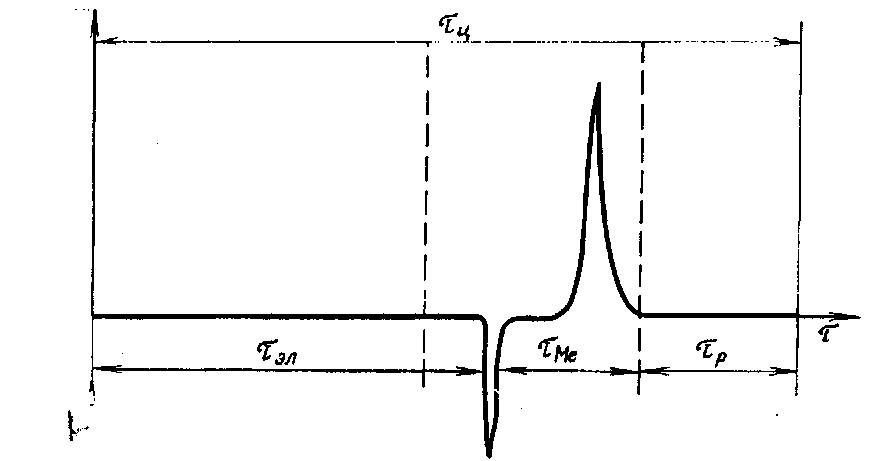
В качестве рабочего электрода применяют серебряный электрод, покрытый пленкой ртути (амальгамированный). Электроды наготавливают из серебряной проволоки диаметром 1...2 мм, закреплённой эпоксидной смолой и полиэтиленполиамином в стеклянной трубке так, чтобы снаружи оставался участок длиной 4...5 мм. Электрод обрабатывают концентрированной НNОз, 10%-м раствором NH4OH, промывают дистиллированной водой и покрывают пленкой ртути, погружая в очищенную ртуть. Обновляют ртутную пленку один раз в месяц. В качестве рабочих электродов можно также использовать электрод в виде висячей ртутной капли, различные разновидности графитовых электродов и электроды из благородных металлов (Ag, Au, Pt).
Ячейка представляет собой стеклянный стаканчик рабочей емкостью 10...20 мл. В процессе концентрирования раствор перемешивают магнитной мешалкой. За 15 с до окончания концентрирования перемешивание прекращают. Переходное время электрохимического растворения регистрируют в успокоенных растворах.
При постоянных условиях концентрирования и растворения величина переходного времени прямо пропорциональна концентрации определяемого элемента в растворе. В качестве примера на рисунке 8 приведены графики зависимости времени перехода ионов меди от ее содержания в растворе.
Выбор состава основного электролита (фона). При определениях методом инверсионной хронопотенциометрии к исследуемым растворам добавляют минеральные кислоты, основания или соли, которые называют основными электролитами, или фонами.
|
Рис. 8. Осциллограммы производных (dE/dτ) кривых растворения меди; условия накопления: фон—1 М раствор NH4Cl +NH4OH, tэл=2 мин; Е,л = -1 В; R = 70 кОм; C1—С7, — концентрация меди (г-ион/л); 1 — калибровочная кривая; 2...8 — графики зависимости времени растворения меди от ее концентрации; τCu — переходное время растворения меди |
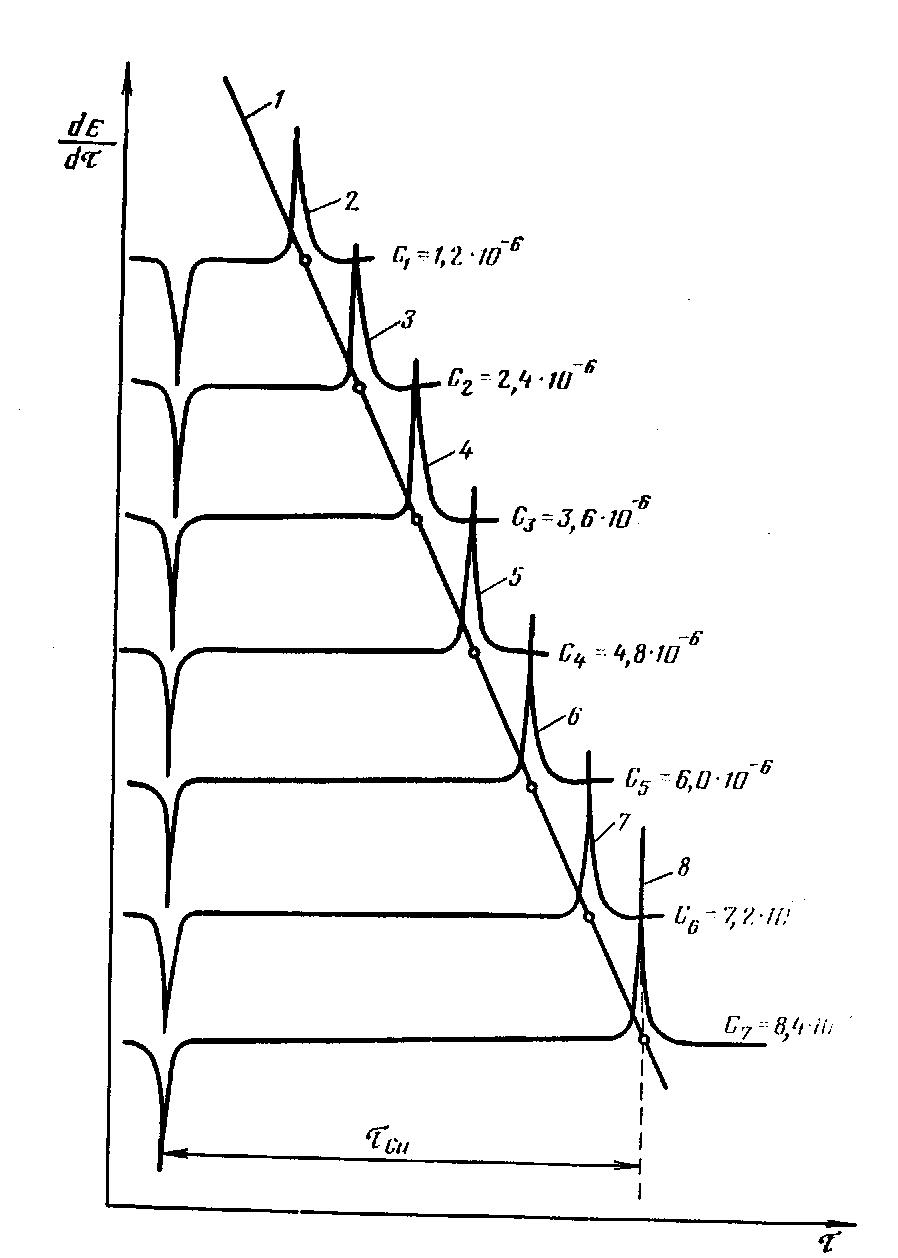
При выборе основного электролита необходимо учитывать:
концентрацию электропроводящих частиц в растворе — она должна быть достаточной, для того чтобы не было большого падения напряжения на сопротивлении, подавлялся миграционный ток, и сохранялось постоянство коэффициентов активности;
чистоту компонентов основного электролита — они должны быть достаточно чистыми или легко очищаться от примесей доступными способами;
избирательность основного электролита — чтобы она была достаточно высокой, к основному электролиту прибавляют комплексообразующие вещества, так как в зависимости от концентрации этих веществ и их химической природы меняется потенциал концентрирования исследуемого иона;
возможное влияние на процесс концентрирования поверхностно-активных веществ — обычно с увеличением концентрации поверхностно-активных веществ уменьшается чувствительность определений.
Выбор потенциала концентрирования. Теоретически потенциал электролиза можно определить, зная соответствующий потенциал полуволны Е0,5 определяемого иона в данной среде. Однако потенциал полуволны, согласно формуле Нернста, приблизительно равен равновесному потенциалу, если концентрации в растворе и амальгаме равны. При инверсионном определении в конце процесса концентрация металла в амальгаме в 100..1000 раз выше, чем в растворе, поэтому потенциал электролиза Еэл должен быть приблизительно на 0,2/n В отрицательнее потенциала полуволны.
На практике Еэл выбирают следующим образом. По таблицам определяют потенциал полуволны восстановления исследуемого иона на соответствующем фоне и устанавливают потенциал электролиза на 0,3...0,4 В отрицательнее. Например, если определение цинка ведут на фоне 1 М КС1,ч потенциал полуволны цинка на этом фоне —1,022 В. Следовательно, для инверсионного определения цинка его концентрирование. необходимо осуществлять при —1,3...1,4 В.
Следует учитывать, что потенциал полуволны существенно зависит от состава фона, поэтому, пользуясь таблицами потенциалов полуволн, нужно обязательно учитывать состав анализируемого раствора. Если исследуемый раствор содержит несколько элементов или требуется определить более электроположительные элементы в присутствии менее электроположительных, можно подобрать потенциал электролиза таким образом, что будет достигнуто эффективное разделение.
Например, анализируемый раствор содержит Cu2+, .Cd2+ и Zn2+. Соответствующие потенциалы полуволн этих ионов на фоне 1 М раствора NH4Cl+NH4OH равны —0,48, —0,74 и —1,33 В. Если установить потенциал электролиза —1,0 В, то на электроде будут концентрироваться медь и кадмий, цинк в этом случае не влияет на инверсионное определение меди и кадмия. Если потенциал электролиза установить —0,6 В, то на электроде будет накапливаться только медь, а кадмий и цинк не окажут влияния на ее определение. При анализе сложных многокомпонентных систем потенциал электролиза определяемого иона устанавливают по экспериментальной зависимости потенциал электролиза — переходное время растворения определяемого компонента
Выбор времени концентрирования. Чувствительность определения методом инверсионной хронопотенциометрии линейно возрастает с увеличением времени концентрирования TЭл. В процессе концентрирования концентрация определяемого вещества в растворе не должна заметно уменьшаться. Обычно таким требованиям отвечает время электролиза меньше 30 мин, а объем раствора больше 10 мл.
При обычных измерениях на ртутных пленочных электродах в области концентрации определяемого иона 10-6...10-7 г-ион/л продолжительность концентрирования составляет 1...10 мин при интенсивном перемешиваний раствора магнитной мешалкой. Вместо перемешивания раствора возможно вращать рабочий электрод.
При анализе более разбавленных растворов время электролиза увеличивают.
Выбор условий растворения. Накопление элемента на рабочем электроде осуществляется в успокоенном растворе при заданном сопротивлении R. Переходное время растворения прямо пропорционально зависит от R. Таким образом, увеличение сопротивления в цепи растворения ведет к увеличению чувствительности определении. Оптимальный диапазон сопротивлений составляет 50...150 кОм.
Для получения устойчивых результатов условия концентрирования и растворения определенного элемента подбирают таким образом, чтобы значения переходного времени электрохимического растворения не выходили за пределы 3...10 с.
Качественные и количественные определения. При исследовании неизвестных растворов для качественной характеристики определяемого элемента пользуются заранее достроенными калибровочными графиками времени появления пика (n) (см. рис. 8) для каждого иона с учетом состава фона. По значению переходного времени τ определяют концентрацию исследуемого элемента. Расчет концентрации выполняют по методу добавок или по калибровочному графику.
Величины пиков с помощью калибровочного устройства осциллографа выражают в милливольтах. По величине потенциала лика при постоянном фоне для любого значения т можно установить определяемый элемент.
Основное условие качественных и количественных измерений при инверсионной хронопотенциометрии — строгая стандартизация условий электрохимического концентрирования элемента на рабочем электроде, а затем электрохимического растворения. При этом переходное время окисления прямо пропорционально концентрации определяемого элемента:
τ = КС
где К - константа
Таким образом, количественный инверсионно-хронопотенцио-метрический метод позволяет применять градуировочный график или метод добавок. Градуировочный график строят по результатам анализа стандартных растворов в координатах τ = fC. По графику находят концентрацию определяемого вещества в исследуемом растворе, выраженную в тех единицах, которые приняты для стандартов, чаще всего в миллиграммах или микрограммах на 1 мл. Для пересчета на содержание в конкретном объекте учитывают навеску, объем вытяжки и все разбавления Или объемы аликвотных частей. На беи абсцисс можно откладывать непосредственно содержание определяемого элемента в почве, растении и др.
При анализе природных объектов состав анализируемого раствора нельзя точно смоделировать. В этих случаях целесообразно применять метод добавок, исключающий различные неконтролируемые отклонения результатов анализа. В исследуемом растворе определяют переходное время определяемого элемента, а затем вводят типовой раствор чистого соединения определяемого элемента, добиваясь увеличения переходного времени примерно вдвое по сравнению с переходным временем исходной пробы.
Приемы пересчета концентрации раствора на содержание элемента в пробе — общие для всех методов количественного анализа.
Чувствительность инверсионно-хронопотенциометрических определений позволяет анализировать растворы с концентрацией определяемого компонента до 5...10-8 моль/л.
При помощи этого метода можно определять макроколичества Си, Pb, Zn, Mn, Fe, Co, Ni, Cd, TI, Sn, Bi и других химических элементов.
Хронополярограф — специализированное устройство, предназначенное для автоматического определения в цифровой форме концентрации металлов. Он представляет собой компактный прибор с встроенной магнитной мешалкой и клавишным пультом управления. По потенциалу электролиза выбирают металл, концентрацию которого необходимо измерить, нажимают соответствующую клавишу, устанавливают время и режим работы электролиза и запускают прибор. Последующие аналитические операции полностью автоматизированы, что обеспечивается микропроцессором. Результат измерения выдается на табло прибора.
При выполнении массовых анализов в память прибора предварительно вводят характеристику «концентрация—длительность». При отсутствии ее используют стандартный раствор. Величину концентрации металла в этом растворе вводят в память прибора, после чего характеристику определяют автоматически.
Полное время, затраченное на измерение концентрации одного металла, составляет 1,5... 10,5 мин. С уменьшением концентрации увеличивается время накопления элемента на электроде