

Физико-химические методы разделения и концентрирования
Экстракция
Значение методов разделения в физико-химических методах анализа трудно переоценить. Перед количественным определением элементного состава образца очень часто выделяют некоторые или все его компоненты.
Основы метода. Многие органические жидкости не смешиваются с водой. При добавлении такой жидкости к воде образуется два слоя. Если плотность органической жидкости больше плотности воды, органический слой находится внизу, а если плотность органической жидкости меньше плотности воды, эта жидкость располагается вверху.
Предположим, что водный раствор, содержащий два растворимых вещества А и В, энергично встряхивают с несмешивающейся органической жидкостью и оставляют смесь до полного расслаивания. Если сродство органического растворителя к одному из растворенных веществ намного больше, чем сродство воды к нему, это вещество полностью перейдет из водной фазы в органическую, или, другими словами, это вещество экстрагируется. Вещество, которое остается в водной фазе, не экстрагируется. Если экстракцию ведут в делительной воронке, нижний слой жидкости можно аккуратно слить и таким образом разделить физическим методом два вещества.
Экстрагирующееся вещество должно хорошо растворяться в органической жидкости, используемой для экстракции. Органический растворитель подбирают таким образом, чтобы после встряхивания с водным раствором капли этого растворителя быстро соединялись между собой и образовывали отдельный слой. Такое быстрое разделение возможно при условии, что отношение плотности органической жидкости к плотности воды, или относительная плотность, должно быть значительно больше или меньше единицы.
Для экстракции органических веществ и соединений металлов в качестве тяжелого растворителя широко используют хлороформ СНСl3. Этому растворителю отдают предпочтение перед четыреххлористым углеродом СС14. Типичными легкими растворителями служат бензол С6Н6 и диэтиловый эфир С2Н5ОС2Н5. Хорошим растворителем для многих экстракционных разделений считают метилизобутилкетон СНзСОСН2СН(СН3)2. Несмотря на то, что плотность трибутилфосфата (С4Н9)3РО4 близка к плотности воды, этот растворитель часто используют для экстракции многих комплексных соединений металлов, причем с большим успехом, чем другие растворители. Для лучшего разделения фаз и повышения селективности экстракции трибутилфосфат иногда предварительно смешивают с бензолом или керосином.
При встряхивании растворенного в воде вещества с несмешивающимся с водой растворителем первое может оказаться как в водной, так и в органической фазе. При равновесии между водой и таким растворителем, как бензол, молекулярные органические вещества переходят в бензол, а ионизированные органические соединения и неорганические соли остаются в основном в водной фазе. Иногда экстракция зависит от рН водного раствора. Например, экстракция фенола обусловлена наличием двух последовательных равновесий:
С
 6Н6ОН
С6Н5ОН
NaOH
С6Н5ОН-
Na+
6Н6ОН
С6Н5ОН
NaOH
С6Н5ОН-
Na+
В нейтральном или кислом растворе фенол находится преимущественно в молекулярной форме и экстрагируется бензолом. Однако в сильном щелочном водном растворе фенол присутствует :в виде фенолят-иона, который не экстрагируется бензолом.
Экстрагирование (экстракция)—процесс разделения смеси жидких или твердых веществ с помощью избирательных (селективных) растворителем (экстрагентов). Физическая сущность экстракции заключается в переходе извлекаемого экстрагируемого вещества из жидкой или твердой фазы в фазу жидкого экстрагента при их взаимном соприкосновении.
Экстракция включает следующие основные операции: смешение исходной смеси веществ с экстрагентом; разделение образующихся двух фаз; удаление и регенерация экстрагента. Применяемый экстракт и исходный раствор должны быть нерастворимы один в другом. Кроме того, экстрагент должен обладать селективностью, возможно большим отличием от исходного раствора по плотности и низкой вязкостью (для облегчения расслаивания фаз), легкой регенерируемостью, химической инертностью, нетоксичностью и доступностью.
Полнота экстракции. Процесс экстракции подчиняется законам диффузии и равновесного распределения. Отношение концентрации экстрагируемого вещества в равновесном экстракте и рафинате называют коэффициентом распределения (D).
Экстракция основана на законе распределения Нернста. При постоянной температуре соотношение концентраций вещества, распределившегося между двумя несмешивающимися жидкостями — фазами, является величиной постоянной:
D = а/b,
где a-—концентрация в органическом растворителе (экстракте); b —концентрация в воде (рафинате). Условие равновесного распределения компонента между двумя фазами при постоянных температурах и давлении — это равенство активностей в каждой фазе.
В практических процессах экстракционный линейный закон распределения часто нарушается, так как различные факторы (например, диссоциация и ассоциация) влияют на активность в растворе и величина коэффициента распределения (D) оказывается функцией концентрации, температуры, рН и др.
Коэффициенты распределения различных классов соединений по-разному зависят от концентраций кислот, рН раствора, растворителя и других условий эксперимента, что позволяет избирательно разделять элементы. Если, например, ионы металлов не экстрагируются, то для экстракции широко используют соли этих металлов с органическими кислотами, их комплексные соединения и др.
Широкому распространению экстракционного метода разделения и концентрации способствует возможность совместить разделение с одновременным определением концентрации в экстракте. Очень часто экстрагируемое соединение окрашено и оптическая плотность пропорциональна концентрации определяемого вещества. Процесс и законы экстракции лежат в основе распределительного хроматографического анализа.
В агрохимии и
почвоведении широко применяют экстракцию
для концентрирования и разделения
микроэлементов, пестицидов и др. Поскольку
концентрацию выражают в массе вещества,
приходящейся на определенный объем
растворителя, можно за![]() писать
уравнение следующим образом:
писать
уравнение следующим образом:
DС = |
Aорг(ммоль)/Vвод |
= |
A (ммоль)Vорг |
Aвод (ммоль)/Vорг |
A орг (ммоль)Vвод |
Если обозначить соотношение количеств растворенного вещества в двух фазах как массовый коэффициент распределения:
DС = |
Aорг(ммоль) |
Aвод (ммоль) |
то уравнение (2) примет следующий вид:
DС = DМ |
Vвод |
Vорг |
DМ = DС |
Vорг |
Vвод |
Если, как это часто бывает при экстракции, Vорг = Vвод, DМ и DС равны между собой, величину DС во всех уравнениях можно заменить на DМ. Если Vорг ≠ Vвод, то величину DМ рассчитывают из значений DС, Vорг и Vвод.
Используя следующее уравнение, можно рассчитать массу вещества (ƒ), оставшуюся в водной фазе:
ƒ = |
Aвод (ммоль) |
= |
1 |
Aорг(ммоль) + Aвод (ммоль) |
DМ + 1 |
Часто в результате однократной экстракции вещество извлекается не полностью. Поэтому после первой экстракции органическую фазу отделяют, а водный раствор встряхивают с новой порцией органического растворителя. Эту процедуру можно повторить несколько раз. Часть вещества, оставшаяся в водной фазе после n экстракций новыми порциями растворителя, равна:
ƒ = |
1 |
(DМ + 1)n |
Вычитая из единицы часть вещества, оставшуюся после экстракции, и умножая разность на 100, получаем процент экстракции после n экстракций:
Е = 100 [1 – |
1 |
] |
(DМ + 1)n |
Считают, что разделение выполнено количественно, если процент экстракции равен 99,9. Если при каждой экстракции процент извлечения равен по крайней мере 90 (D=9), для количественного выделения желаемого вещества необходимо провести двух- или трехкратную экстракцию новыми порциями растворителя. Если коэффициент распределения невелик, для полного выделения вещества необходимо проводить большее количество экстракций со свежими порциями растворителя. Выделение таких веществ удобно вести в экстракторе непрерывного действия.
Широко применяют метод экстракции для определения жиров, пигментов, пестицидов и продуктов их распада. Определение этих веществ после их концентрирования методом экстракции существенно облегчается, повышается точность метода, чувствительность.
Хроматография
Теоретические основы. В 1903 г. М.С. Цвет впервые изложил принципы хроматографии (греч. «хромо» — цвет, «графо — пишу) и создал метод разделения пигментов зеленых растений. Для хроматографического анализа нужно очень малое количество веществ — десятые доли миллиграмма или даже микрограммы и доли микрограмма. Важное преимущество хроматографии перед другими методами разделения вещества в том, что при его применении вещества не подвергаются химическим изменениям. Хроматографический метод позволяет разделять и анализировать сложные смеси, очищать вещества от ненужных примесей, концентрировать и идентифицировать их.
Хроматографическое разделение веществ происходит под влиянием различной адсорбируемости компонентов смеси, различного распределения компонентов смеси между двумя несмешивающимися жидкими фазами и различной растворимостью образующихся осадков. Различают четыре вида хроматографии: адсорбционную, ионообменную, распределительную, осадочную.
Адсорбционная хроматография основана на различной способности компонентов к адсорбции (избирательная адсорбция) на том или ином сорбенте. Адсорбция и десорбция веществ в колонке происходит под действием межмолекулярных сил.
Ионообменная хроматография основана на обменной адсорбции, т. е. ионы, содержащиеся в хроматографируемом растворе, обмениваются на эквивалентное количество подвижных ионов, входящих в состав ионообменника. Хроматограммы при этом образуются в результате различной способности к обмену попон хроматографируемого раствора. Реакция ионного обмена обратима.
Распределительная хроматография основана на распределении растворенных веществ между двумя несмешивающнмнся растворителями. Следовательно, в распределительной хроматографии используют различия в коэффициентах распределения хроматографируемых веществ между двумя несмешивающимися жидкостями — подвижным и неподвижным растворителями. Различие коэффициентов распределения определяет неодинаковую скорость движения компонентов смеси, поэтому в конечном итоге образуется хроматограмма, состоящая из отдельных зон компонентов смеси.
Осадочная хроматография основана на принципе последовательного осаждения малорастворимых соединений. При пропускании анализируемого раствора через носитель, смешанный с соответствующим осадителем, образуется осадочная хроматограмма, причем пространственное размещение образующихся осадков сверху вниз по колонке происходит в порядке увеличения их растворимости.
Классификация хроматографических методов по механизму элементарного процесса, агрегатному состоянию систем, в которых производится разделение, и по характеру осуществления процесса можно представить в виде следующей схемы:
Характерная особенность всех видов хроматографии — многократность повторения элементарных процессов, например сорбции-десорбции, экстракции-реэкстракции и др. Это обусловливает высокую эффективность хроматографических методов для разделения близких по химическим свойствам элементов.
|
|
|
|
Хромотография |
|
|
||||||
|
|
|
|
|
|
|
|
|
||||
По агрегатному состоянию |
|
Газовая |
|
Жидкостная |
|
Газово-жидкостная |
|
|||||
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
||||
По механизму разделения |
|
Адсорбционная |
|
Распределения |
|
Ионообменная |
|
Осадочная, окислительно-востановительная,адсорбционно-комплексообразовательная |
||||
|
|
|
|
|
|
|
|
|
||||
По способу ведения процесса |
|
Колоночная |
|
Капиллярная |
|
Плоскостная (бумажная, тонкослойная) |
||||||




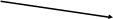

Количественная, бумажная и тонкослойная хроматография. Бумажную и тонкослойную хроматографию можно использовать для количественного анализа смесей. Для этого на стартовую линию наносят в виде пятна или полосы точный объем раствора пробы при известной концентрации. Нанесение раствора осуществляют микропипетками, микрошприцами или специальными приспособлениями. При нанесении пробы стремятся, чтобы в ней содержалось не менее 20... 500 мкг определяемого вещества (в зависимости от методики количественного определения).
Затем хроматографируют и количественно определяют вещества в полученных пятнах или полосах. Используют несколько способов определения компонентов пробы: по площади зон, полученных на хроматограмме; по измерению физических и физико-химических свойств зон на хроматограмме; экстрагированием зоны соответствующим растворителем и анализом экстракта.
Определение по площади зон основано на явлении насыщения слоя адсорбента или бумаги веществом. Каждый тип адсорбента имеет определенную емкость и не может адсорбировать в единице массы больше определенного количества вещества. При хроматографировании неадсорбированная часть вещества переходит в подвижную фазу и поступает на свободные участки адсорбента. Вещество распределяется на хроматограмме на площади, пропорциональной его массе (рис. 1, а). Зависимость между массой вещества q и площадью пятна S на хроматограммах представляет собой логарифмическую функцию:
![]() = a·lg
+ b,
где а и b
— коэффициенты.
= a·lg
+ b,
где а и b
— коэффициенты.
Эта зависимость линейна для количества вещества от 1 до 80... 100 мкг.
При таком методе для исключения больших ошибок применяют стандартные растворы, определенные объемы которых с помощью микропипетки наносят на хроматограмму, стремясь получить серию пятен с различным содержанием стандарта. На эту же хроматограмму наносят определенный объем раствора пробы. Осуществляют хроматографирование, подсушивают хроматограмму, обрабатывают ее раствором реактива, подсушивают снова и измеряют площадь зон.
Измерение площади ведут планиметром. При отсутствии планиметра под хроматограмму подкладывают копировальную и миллиметровую бумагу, обводят по периметру пятно зоны карандашом и подсчитывают площадь (мм2) изображений пятен на миллиметровой бумаге. Строят калибровочный график зависимости площади зон от концентрации стандарта (рис. 1,6) и по графику определяют массу компонента в растворе пробы. Ошибка метода измерения площади зон достигает 5—10%, поэтому метод используют для ориентировочных анализов.
|
Рис. 1. Зависимость площади пятен на хроматограмме от количества вещества: а — хроматограмма; б — калибровочный график: S — площадь пятна на хроматограмме; С — концентрация |
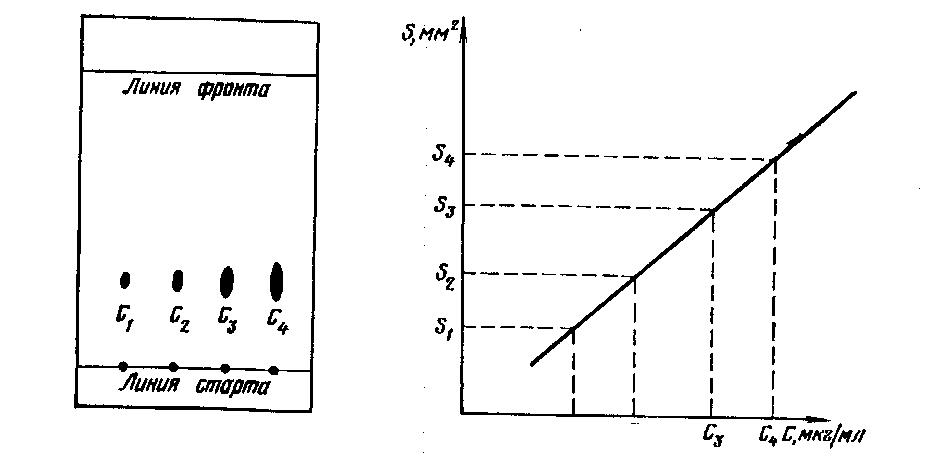
Более точен денситометрический метод определения веществ на хроматограммах (ошибка 1—2%). При денситометрии выполняют измерение оптического поглощения проявленной хроматограммы сканирующим лучом в проходящем или отраженном свете на специальных приборах — денситометрах (рис. 2). На денситограмме получают пики, площадь которых пропорциональна содержанию вещества в пятне. Построив с помощью стандартов калибровочный график, измеряют площадь пика компонента и по графику определяют его массу в пробе.
Получает развитие также спектрофотоденситометрическое и флуориметрическое определение веществ на хроматограммах, В первом случае используют специальные спектрофотоденситометры, измеряющие поглощение вещества в монохроматическом свете, во втором измеряют флуоресценцию пятна при облучении хроматограммы УФ-светом.
Широко распространен способ экстрагирования компонентов из зон подходящим растворителем. При таком способе на хроматограмму наносят стандартный раствор и раствор пробы. После получения хроматограммы ее обрабатывают, детектируя зону стандарта: вырезают часть хроматограммы с зоной компонента пробы и экстрагируют его подходящим растворителем. Полученный раствор анализируют инструментальным методом, имеющим высокую чувствительность. Чаще всего применяют спектрофотометрические и фотоколориметрические методы. Если вещество бесцветное или не обладает поглощением в УФ-области, с экстрактом выполняют фотометрическую реакцию, позволяющую получить интенсивно поглощающее производное вещество.
|
|
Рис. 2. Схема денситометра: 1 - протяжный механизм хроматограммы; 2 — источник света; 3 — хроматограмма; 4— самописец; 5 — усилитель; 6 — фотоэлемент; 7 — объектив |
Рис. 3. Двухмерные хроматограммы аминокислот в тонком слое сили-кагеля: а — аминокислот-свидетелей; б — вытяжки из свежего мяса; в — вытяжки из мяса сомнительной свежести; г — вытяжки из несвежего мяса: 1— фенилаланин; 2 — лейцин; 3 — метионин; 4 —тирозин; 5 —валин; 6 — аланин; 7 — пролин; 8 — глицин; 9 — серин; 10 — гистидин; 11—глутаминовая кислота; 12 — аспарагиновая кислота; 13 — аргинин+лизин |
Для разделения смесей веществ, которые слабо разделяются при хроматографировании, используют метод разгонки по принципу двухмерной хроматографии, когда осуществляют двухмерное хроматографирование (в направлении «А» и «В», перпендикулярных один другому). При этом разгонку в направлении «А» выполняют с одним подвижным растворителем, а после сушки разгон ведут в направлении «В» с другим растворителем или растворителем, имеющим иную величину рН. Пример разгонки аминокислот по принципу двухмерной хроматографии представлен на рисунке 3.
Бумажная и тонкослойная количественные хроматография имеют важное преимущество по сравнению со многими другими методами анализа — высокую чувствительность. Этими методами можно определить 10—20 мкг вещества с точностью до 5—• 7%.
Газовая хроматография. Наиболее важные хроматографические методы — газо-адсорбционная и газо-жидкостная хроматография. При газовой хроматографин происходит распределение компонентов анализируемой смеси между газообразной и твердой или жидкой фазами. В установке для газовой хроматографии используют твердый инертный пористый носитель, в газо-жидкостной хроматографии он покрыт тонким слоем жидкой фазы. Жидкая или твердая фазы неподвижны. Подвижной фазой служит газ-носитель, в котором содержится анализируемая проба.
При выполнении газовой хроматографии в нагретый до определенной температуры поток раза-носителя вводят анализируемую пробу. Вещества пробы испаряются и вместе с потоком газа поступают в термостатированную колонку с неподвижной фазой (адсорбентом). В колонке протекают многократные процессы адсорбции и десорбции на твердом носителе или растворения и выделения в жидкой пленке смеси газообразных веществ. Разделение сложной смеси здесь зависит от коэффициентов адсорбции или распределения анализируемых веществ между фазами. На выходе из .колонки смесь разделяется на индивидуальные вещества, поступающие с потоком газа на детектор.
Любой газовый хроматограф (рис. 4) состоит из источника постоянного потока газа-носителя 1, регулятора потока газа 2, дозирующего устройства для количественного ввода анализируемой пробы 3, термостатированной хроматографической колонки 4, детектора 5, самописца 6, блока нагрева колонки 7 и необходимого в некоторых случаях устройства для улавливания компонентов смеси после их разделения.
|
Рис. 4. Схема газового хроматографа: 1 — источник постоянного потока газа-носителя; 2 — регулятор потока газа; 3 — дозирующее устройство для количественного ввода анализируемой пробы; 4 — термостатированная хроматографическая колонка; 5 —детектор; 6 — самописец; 7 — блок нагрева колонки. |
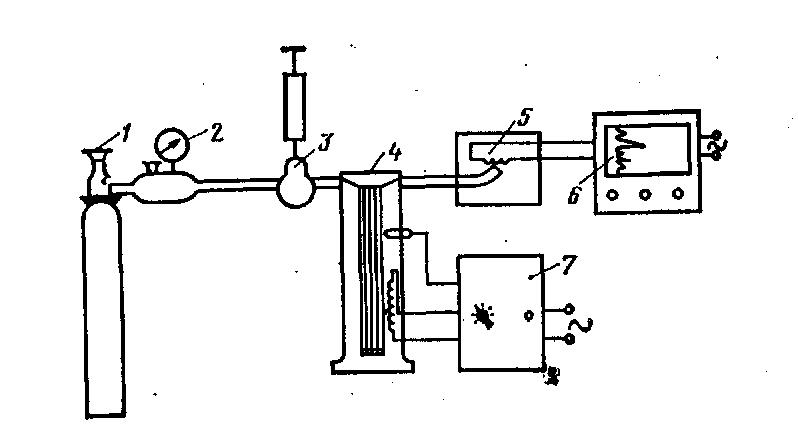
Газ-носитель подается из газового баллона через редуктор. Расход газа-носителя определяют специальными расходомерами-ротаметрами. Для очистки газа от влаги и других примесей используют склянки или V-образные трубки, заполненные хлоридом кальция, силикагелем. Их устанавливают перед дозатором. Дозируют и вводят пробу в хроматографы дозаторами. В лабораторной практике используют специальные шприцы. Для введения проб газа большого объема пользуются бюретками и отсекающими петлями. Хроматограф имеет приспособления, обеспечивающие смешение пробы с газом-носителем или ее испарение. Поток газа-носителя вместе с пробой поступает в колонку. В газовой хроматографии применяют термостатированные прямые, U-образные и спиральные колонки диаметром от 2 до 12... 15 мм, длиной 2... 20 м. Колонки изготавливают из стекла, меди, латуни, стали. Очень большое значение имеет полное и равномерное наполнение колонки сорбентом и постоянство температуры, которых достигают термостатированием колонки.
Детектор — наиболее важный узел газового хроматографа. Он реагирует на изменение состава газа при выходе и передает эти данные регистрирующему прибору. Детекторы подразделяют на интегральные и дифференциальные. Сигнал интегрального детектора пропорционален общей массе вещества в потоке газа. При прохождении через детектор чистого газа-носителя на ленте записывается горизонтальная линия, если через детектор проходит компонент смеси, перо самописца перемещается, вычерчивая ступень. Таким образом, хроматограмма, полученная с помощью интегрального детектора, состоит из ступеней, высота которых пропорциональна массе компонента, соответствующего данной ступени.
В хроматографах чаще используют дифференциальные детекторы, регистрирующие разностный сигнал. Работа дифференциальных детекторов основана: на различии в теплопроводности
ходимостью анализа 1ВЫСОКОКИПЯЩНХ (>4ФО°С) или неустойчивых соединений, которые не разделяются методом газовой хроматографии, во-вторых, необходимостью увеличить скорость разделения и повысить эффективность метода колоночной жидкостной хроматографин. Для этогЪ применяли колонки ic малым внутренним диаметром (2...6 мм); для ускорения массообмена уменьшили диаметр частиц сорбента (5...50 мкм), что, в свою очередь, привело к необходимости увеличить давление на 'выходе колонки до 0,5...40 МП а.
Выпускаемые промышленностью жидкостные хроматографы снабжены высокочувствительными детекторами, позволяющими определять до 10~9..ЛО~10 г вещества. Детекторы в жидкостных хроматографах можно объединить ib следующие группы: оптические детекторы, составляющие около 92 % всех применяемых детекторов (абсорбционные, люминесцентные, рефрактометры); электрохимические детекторы (потенциометрмчеокие, амперомет-рические и др.); другие детекторы (транспортные, газовые, мнк-роадсорбцшшные), например 'проточные рефрактометры, регистрирующие изменения показателя преломления растворов с чувствительностью порядка 10~7 г/мл, спектрофотометры с фиксированными ('254 и 365 нм) >н регулируемыми полосами поглощения света в ультрафиолетовой и 'Видимой областях спектра поглощения и спектрофлюориметры с чувствительностью, достигающей 10~9... 10-!0 г/мл.
Современные детектирующие устройства в жидкостной хроматографии обеспечивают непрерывную регистрацию (концентрации анализируемых веществ ib вытекающем из колонки потоке на уровне 10"2...10-4 %.
В жидкостной хроматографа и длина используемых колонок меньше, чем в газовой, и составляет 10...25 см. Частицы сорбента имеют размеры 5...10 мкм и.строго одинаковы .по размеру. В качестве сорбентов в адсорбционно-жидкостной хроматогра-фии применяют силикагелн, оксид алюминия, оксид магния, сахарозу, полимеры и др.
Обычно на'несение пленки на (поверхность твердого носителя не позволяло получить устойчивый сорбент (пленка легко смывалась органическими растворителями). Выход был найден, когда гндроконльные группы поверхности силикагеля стали заменять на специфичные группы, например нитрильные, кислотные, силильные и т. д.
Адсорбционно-жидкостный и жидко-жидкостный методы хроматографии очень тесно 'переплелись между собой \и имеют общую область применения для разделения смесей нуклеотидов, витаминов, лекарственных препаратов, сложных биологически активных смесей, органических соединений и др.
Достаточно высокая скорость анализа, низкий предел обнару-
Жения, высокая эффективность колонки, возможность определять любые вещества (кроме газов) привели к быстрому развитию ВЭЖХ.
В основе молекулярно-ситовой (гель-фильтрационной, гель-) хроматографии лежит принцип разделения смеси веществ по их молекулярным размерам или молекулярным массам. Такие фи* зико-химические свойства веществ, как сродство к сорбенту, растворимость, поверхностный разряд, играющие важную роль 1Э .других вариантах хроматографии, здесь, как правило, не имеют значения, так 'как разделяемые вещества практически не взаимодействуют с носителем.
В качестве носителей в молекулярно-ситовой хроматографии используют различные гели с трехмерной сетчатой структурой: декстрановые, шолиакриламидные, агарозные, гели на основе стирола и дивинил бензол а, пористые силикагели, а также -пористые стеклянные гранулы. Все гели и стекла имеют поры строго определенных размеров, по возможности we содержат ионоген-ных <rpyimi и не обладают химическим или биологическим сродством к анализируемым веществам.
Разделение смеси веществ происходит за счет того, что в поры геля могут диффундировать только вещества, размеры .молекул которых не превышают размеров пор. Объем растворителя внутри гранул геля принято называть внутренним объемом колонки. В результате диффузии внутрь пор геля молекулы меньшего размера проходят больший путь и элюируются с геля позже, чем более крупные молекулы, не проникающие в поры геля. 'Последние проходят между .гранулами геля и выходят из колонки с растворителем в так называемом свободном, ияи внешнем, объеме колонки, т. е. в .объеме растворителя, распределенного между гранулами геля и составляющего меньшую часть от общего объема колонки.
В молекулярно-ситовой хроматографии, как и в любом другом виде хроматографии, решающим условием хорошего разделения смесей веществ служат правильный выбор носителя и его упаковка в колонке. Носитель выбирают с таким расчетом, чтобы основная масса разделяемых веществ могла проникнуть внутрь пор геля. Качество же набивки колонки можно проверить с помощью высокомолекулярного окрашенного полисахарида —синего декстрана, который должен продвигаться к колонке с гелем в виде резко очерченной и строго горизонтальной зоны. Характер искривления зоны указывает на ошибки, допущенные в процессе заполнения колонки гелем (слишком густой или слишком жидкий гель, неправильная скорость подачи растворителя в колонку, невертикальная установка колонки и др.).
Особенно интенсивное развитие молекулярно-ситовая хрома-тографня получила в последние два десятилетия в связи с вне-
Дрёнием & Химическую н биохимическую практику сёфадексов — декстрановых гелей, поперечно сшитых эпихлоргидрином. На различных типах сефадексов можно фракционировать разнообразные химические вещества с молекулярными массами 700 000...800 000 Дальтон, поэтому их широко используют для выделения и очистки биополимеров — белков, пептидов, поли- н олнгосахаридов, нуклеиновых кислот и др.
Хемосорбционная хроматография — это группа хроматогра-фическнх методов. К ней относятся варианты жидкостной хрома-гографии, для которых характерно проявление относительно сильных взаимодействий между сорбентом и сорбатом.
Хемосорбцнонная хроматография широко используется для качественного и количественного определения различных неорганических веществ, их разделения, концентрнрования и выделения.
Осадочная хроматография впервые предложена советскими исследователями Е. Н. Гапон и Т. Б. Гапон в 1948 г. В отличие от рассмотренных выше для данного метода характерно многократное повторение элементарных химических актов образования и растворения осадков и их закрепление в порах носителя (в месте образования) в процессе фильтрации раствора разделяемых веществ через колонку, содержащую осадитель.
Различия в растворимости образующихся в колонке осадков (при взаимодействии раствора разделяемых веществ с осадите-лем) обусловливают порядок распределения осаждаемых веществ в зонах колонки, а следовательно, и их разделение. Образование осадков разделяемых веществ в хроматографическон колонке происходит только при достижении их произведений растворимости.
В насыщенном растворе малорастворимон соли электролита КА между осадком КА и ионами К+ и А~ в растворе устанавливается динамическое равновесие:
КА*=*К+ + А~. (118)
Произведение активностей .ионов малорастворимого соединения (электролита) в его насыщенном растворе при данной температуре— величина постоянная (правило произведения растворимости), т. е. ПРКА=ак**аА~* где ак+ и аА~ — активности катиона и аниона соответственно. Выпадение малорастворнмого соединения в осадок происходит в пересыщенном растворе, т. е. тогда, когда
«к+-«А->ПРКА. О* 9)
Порядок распределения осадков двух и более хроматографи-руемых ионов сверху вниз по колонке или хроматографнческон бумаге определяется отношениями значений произведений рас-
твори мости осадков, отношениями молярных концентраций разделяемых ионов и их валентностью.
Например, если через колонку с оксидом алюминия, содержащую в качестве осадителя сульфат серебра, фильтровать раствор смеси галогенидов щелочного металла, то в результате обменной реакции образуются осадки галогенидов серебра. Последовательность их распределения на колонке будет точно соответствовать величинам их произведений растворимости: в верхней зоне расположится желтый осадок йодида серебра (ПРАв| = 1,ЫО~16), в средней — голубовато-серый осадок бромида серебра (ПРАввг = 6-10~13) и в нижней —белый осадок хлорида серебра (ПРАес1= 1,8- Ю"10), Часто в Осадочной хроматогра-фии получаются окрашенные зоны с четкими границами, что позволяет проводить количественное разделение анализируемых компонентов в растворе.
Метод окислительно-восстановительной хроматографии (ре-докс-хроматографии) впервые предложен советскими исследователями К- М. Ольшановой и А. Н. Щеколдйной в 1960 г. Как вариант хемосорбционной хроматографии он обладает всеми особенностями, характерными для хроматографического процесса: мгновенным установлением равновесия в окислительно-восстановительной системе, многократностью повторения основного элементарного акта окисления-восстановления и динамичностью процесса.
Методом окислительно-восстановительной хроматографии главным образом разделяют смеси неорганических веществ. Он обусловлен различиями в скоростях окислительно-восстановительных реакций, протекающих между окислителем и восстановителем, содержащимися в составе наполнителя колонки, и'ионами анализируемого раствора, а также удерживанием продуктов реакции в порах носителя на месте их образования.
Окислительно-восстановительные реакции сопровождаются переходом электронов от одних атомов или ионов к другим. Каждая окислительно-восстановительная пара содержит окислительную форму (окислитель), образованную элементом в более высокой степени окисления, и восстановительную форму (восстановитель), образованную элементом в более низкой степени окисления. Следовательно, окисление — это процесс отдачи электронов, а восстановление — процесс присоединения их атомом или ионом.
Направление, скорость и характер протекания окислительно-восстановительной реакции зависят от кислотности среды, температуры, присутствия катализатора и концентрации реагирующих веществ.
Из всех возможных окислительно-восстановительных процессов, протекающих в колонке между окислителем (восстанови-