

-
Физико-химические методы анализа.
-
Хроматография. Сущность метода. Области применения. Применение хроматографии в медицинской химии. Тсх – быстрота, простота и информативность
-
Хроматографический анализ — совокупность методов разделения однородных многокомпонентных смесей, основанных на использовании сорбции в динамических условиях. Это физический метод разделения, при котором разделяемые вещества распределяются между двумя фазами. Одна из фаз неподвижна, другая — подвижна и фильтруется через слой неподвижной фазы. Разделение основано на различиях коэффициентов распределения компонентов смеси между подвижной и неподвижной фазами, это в свою очередь вызывает различия в скорости переноса компонентов по длине неподвижной фазы. Поток подвижной фазы вызывает дифференцированную миграцию компонентов смеси из первоначальной зоны в пористую сорбционную среду неподвижной фазы.
Пояснения с рисунками.
После хроматографического разделения вещества могут быть количественно определены многими, в том числе неспецифическими методами. Хроматографический анализ — это гибридный метод, сочетающий разделение и детектирование (определение). Разделение и количественное определение часто осуществляют в одном приборе — хроматографе.
Известно много вариантов хроматографического анализа. В зависимости от агрегатного состояния фаз хроматографические методы классифицируют следующим образом.
-
Подвижная фаза — газ — газовая хроматография: газо-адсорбционная или газотвердофазная (неподвижная фаза — твердый сорбент); газожидкостная хроматография (неподвижная фаза — жидкость, нанесенная на инертный носитель).
-
Подвижная фаза — жидкость — жидкостная хроматография: жидко-жидкостная хроматография (неподвижная фаза — жидкость); жидкостно-адсорбционная хроматография (неподвижная фаза — твердый сорбент.)
Газовая хроматография
Это физический способ разделения смесей летучих соединений, основанный на распределении веществ между двумя фазами.
Одна из фаз неподвижна и обладает большой поверхностью, другая, подвижная фаза — инертный газ (газ-носитель), протекающий через колонку с неподвижной фазой. Исследуемый газ или жидкость в виде паров смесь вводят в газ-носитель. Каждый компонент разделяемой смеси движется со своей скоростью. Для контроля потока газа-носителя на выходе из колонки помещают детектор, т.е. прибор, сигнал которого зависит от состава потока газа. Действие детектора основано на измерении одного из физических параметров разделяемых газообразных компонентов. Например, есть детекторы, измеряющие теплопроводность, электропроводность, плотность, показатель преломления света и др. Применяют плазменно-ионизационные, фотоионизационные, акустические, масс-спектрометрические, термохимические и другие детекторы.
Газоадсорбционная хроматография — вариант газовой хроматографии, характеризующийся применением твердой неподвижной фазы (силикагель, активный уголь и др.) и инертного газа-носителя. Разделение определяется адсорбционными свойствами наполнителя колонки по отношению к разделяемым соединениям. По выходе из колонки газ исследуют при помощи детектора.
Газожидкостная хроматография. Неподвижная фаза представляет собой нелетучую жидкость (силиконовое масло, высшие алифатические углеводороды и др.), нанесенную в виде тонкой пленки на твердый инертный носитель (измельченное стекло, керамика, полимер и др.). В колонке поддерживается постоянная высокая температура (изотермическая хроматография). Жидкая или газообразная проба анализируемого вещества испаряется в камере перед колонкой и пары увлекаются потоком инертного газа в колонку. Разные компоненты анализируемой смеси вследствие различной растворимости в жидкой фазе движутся по колонке вместе с инертным газом с разной скоростью и в разное время появляются на выходе из колонки. Далее поток газа проходит через детектор, отмечающий появление примеси в газе-носителе. Показания детектора регистрируют во времени, образуется хроматограмма, состоящая из ряда пиков, каждый пик соответствует одному из компонентов анализируемой смеси. Площадь, занимаемая пиками, служит мерой для определения количества (концентрации) данного компонента.
Можно вводить в пробу известное количество стандартного раствора некоторого летучего вещества и строить график зависимости отношения площадей пиков определяемого и стандартного веществ от отношения масс обоих веществ (метод внутреннего стандарта). Сумму площадей всех пиков принимают за 100% и вычисляют содержание отдельных компонентов (метод нормализации).
Жидкостная хроматография
Это вариант хроматографии, при котором пользуются жидкой подвижной фазой, неподвижная фаза может быть жидкой (жидко-жидкостная хроматография) или твердой (жидкостно-адсорбционная хроматография).
Высокоэффективная жидкостная хроматография, высокоскоростная жидкостная хроматография. Применяют носители, состоящие из тонкого пористого слоя сорбента, окружающего твердое непроницаемое ядро (поверхностно-пористые насадки), помещенного в колонку диаметром 1—3 мм. Скорость прохождения жидкости — 1—5 мл/мин, разделение осуществляется за 1—2 мин [235].
Мультихроматография — разделение компонентов одной и той же пробы при использовании минимум 2 колонок (обычно с полярной и неполярной неподвижной фазой). Реализуется ступенчатое изменение селективности разделительной системы за счет изменения времени пребывания сорбента в контакте с одной из неподвижных фаз. Достигается почти оптимальное разделение смеси [237].
Противоточная хроматография — метод жидко-жидкостной распределительной хроматографии без применения твердого носителя [227].
В зависимости от принципа разделения различают следующие виды хроматографии.
Распределительная (экстракционная) хроматография. Разделение основано на различной растворимости определяемых веществ в подвижной и неподвижной фазах [226].
Бывают :
Распределительная тонкослойная хроматография.
Неподвижная фаза — ундекан, тетрадекан, парафиновое масло, силиконовые масла и др.
Подвижная фаза — полярные органические растворители.
Экстракционная хроматография — вариант жидкостной хроматографии в колонке, сочетание жидкостной экстракции с техникой хроматографии, экстракция становится многостадийным процессом. Неподвижная фаза — органический растворитель или раствор на его основе, например дитизон, дитиокарбаминаты и другие вещества, растворенные в органических растворителях, сорбированных на твердом носителе. Подвижная фаза — анализируемый водный раствор. Многочисленность экстракционных реагентов существенно расширяет методику разделения.
Ионообменная хроматография — разделение компонентов смеси основано на различии констант ионообменного равновесия. В качестве неподвижной фазы используют вещества, способные к обмену ионов (иониты). Поглощение растворенных веществ твердой фазой является результатом межфазных ионообменных реакций.
Элютивная ионообменная хроматография. Колонку с ионообменной смолой вначале обрабатывают раствором того электролита, который будет использован в качестве элюента.
Ионная хроматография — автоматизированный скоростной вариант ионообменной хроматографии.
Гель-хроматография, гель-проникающая хроматография, гель-фильтрационная хроматография, хроматография на молекулярных ситах — вариант хроматографического анализа, основанный на различной доступности пор сорбента для макромолекул разных размеров.
Хроматографические методы по способу разделения классифицируют следующим образом [234].
Вытеснительная хроматография. Сначала в колонку вводят разделяемую смесь (А+Б+В+ и т.д.), затем непрерывно подают раствор вещества Г, обладающего наибольшим сродством к неподвижной фазе — вытесняющий компонент. Он вытесняет все ранее удержанные компоненты и выталкивает их из колонки. Компоненты смеси вытесняют друг друга из неподвижной фазы. Компоненты элюируются из колонки в определенной последовательности. Компонент с наименьшим сродством к неподвижной фазе выходит первым, компонент Г — последним.
Вытеснительная ионообменная хроматография. Сначала смолу в колонке переводят в форму иона, имеющего коэффициент селективности меньший, чем коэффициенты селективности ионов разделяемой смеси. Затем вводят раствор разделяемой смеси (около десятой части обменной емкости колонки). Пропускают раствор элюента, последний содержит ион, имеющий больший коэффициент селективности. Термическая десорбция — вариант вытеснительного анализа. Роль вытеснителя играет нагретая трубчатая печь, надвигающаяся на слой сорбента, полосы разделившихся веществ примыкают друг к другу
Фронтальная хроматография. Разделяемую смесь непрерывно пропускают через колонку с сорбентом. На последнем образуются зоны сорбированных веществ. Наименее сорбирующийся компонент выделяется в чистом виде первым за слоем сорбента. Следующая фракция содержит наряду с этим еще один компонент, занимающий по сорбируемости второе место. Последующие фракции содержат 3, 4 и более компонентов. Наконец выходит порция такого же состава, как и начальная смесь [219, 220, 223].
Проявительная хроматография, проявительный анализ, элюционная хроматография. Анализируемую пробу вводят в колонку с адсорбентом и пропускают газ-носитель (элюент). При этом смесь передвигается по слою сорбента. Скорость движения компонентов смеси в зависимости от их сорбируемости различна. Разделяемые компоненты выделяются из колонки отдельными зонами (полосами), между которыми выходит чистый газ-носитель [219, 220, 223].
По способу экспериментирования различают ряд хроматографических методов.
Колоночная насадочная хроматография. Разделение осуществляется в колонках, которые заполняют сорбентом (насадкой), длина колонки 1 —10 м, внутренний диаметр 3—6 мм [234].
Комплексообразовательная хроматография. Разделение смеси веществ обусловлено различной устойчивостью и сорбируемостью комплексных соединений, образующихся в результате взаимодействия компонентов со специально введенным в систему комплексообразующим реагентом [244].
Редокс-хроматография, окислительно-восстановительная хроматография. Разделение веществ обусловлено разными скоростями окислительно-восстановительных реакций, протекающих между окислителем и восстановителем, содержащимися в колонке, и ионами хроматографируемого раствора. Колонка заполнена окислительно-восстановительным полимером, обладающим также и ионообменными свойствами — редокситы или электро-ионообменники [233].
Электрохроматография. Движение заряженных частиц осуществляется под действием приложенного напряжения. Скорость движения частиц определяется их массой и зарядом [220].
Высаливающая элюентная хроматография — разделение неэлектролитов или слабых электролитов посредством вымывания с колонки водным раствором соли высокой концентрации.
Повторная (многократная) хроматография. Обычно для получения хроматограммы подвижный растворитель пропускают по пластинке (колонке) один раз — однократная хроматография. Однако для лучшего разделения зон проводят многократное хроматографирование. При повторных операциях можно использовать либо тот же, либо иной растворитель.
Капиллярная хроматография — колонкой служит капилляр, его длина 25—100 м, внутренний диаметр 0,1 — 1 мм. Капилляр готовят из стекла, меди, стали, полимеров и др. Внутренняя поверхность капилляра покрыта тонким слоем неподвижной жидкой фазы (например, скваланом). В таких колонках можно разделять близкие по свойствам вещества [222, 247].
Радиогазовая капиллярная хроматография — вариант капиллярной хроматографии для анализа органических веществ, меченных |4С [248].
Бумажная хроматография — один из вариантов хроматографического метода, основан на применении специальных сортов фильтровальной бумаги в качестве среды, в которой происходит разделение смесей.
Двумерная хроматография — вариант бумажной или тонкослойной хроматографии, развитие процесса последовательно в двух взаимно перпендикулярных направлениях. Используют квадратные отрезки фильтровальной бумаги или квадратные пластинки с нанесенным тонким слоем сорбента (размер 20 на 20 см). Сначала хроматографируют в одном направлении, затем высушивают и хроматографируют в направлении, перпендикулярном первому процессу, применяя либо тот же, либо другой растворитель. Двумерную хроматографию применяют для разделения многокомпонентных смесей [238, 244].
Обычную бумажную или тонкослойную хроматографию называют одномерной.
Денситометрия — исследование бумажных или тонкослойных хроматограмм после их проявления по светопоглощению пятен непосредственно на бумаге или пластинке с последующим определением по градуировочному графику.
Тонкослойная хроматография, адсорбционная тонкослойная хроматография — микрометод жидкостной хроматографии, выполняемый на пластинках в тонком слое адсорбента. Высота слоя адсорбента значительно меньше ширины (линейная тонкослойная хроматография). Подвижная фаза перемещается главным образом за счет капиллярных сил. Адсорбенты — силикагель, оксид алюминия, измельченная целлюлоза, смолы и др.
На одну сторону пластинки нанесен тонкий слой измельченного адсорбента. На расстоянии 1 см от края пластинки отмечают стартовую линию, на которую помещают каплю анализируемого раствора. Пластинку ниже стартовой линии погружают в систему растворителей. По мере продвижения жидкости вверх по слою адсорбента происходит разделение смеси веществ (восходящее элюирование). Отмечают границу подъема жидкости (линия фронта растворителя), высушивают и всю поверхность и осматривают ее под ультрафиолетовым светом или смачивают раствором соответствующего реагента (проявление). При этом разделенные вещества обнаруживают по появлению пятен между стартовой линией и фронтом растворителя. Измеряют расстояние от центра пятна до линии старта (отрезок АБ), находят расстояние от линии старта до фронта растворителя (отрезок АВ): АБ/АВ=Rf — качественная характеристика вещества, образовавшего данное пятно.
Для достижения более полного разделения пользуются градиентной тонкослойной хроматографией (градиентное элюирование) — применение элюирующей системы с меняющейся концентрацией солей (градиент концентрации), меняющимся отношением полярных и неполярных растворителей (градиент полярности), меняющимся значением рН (градиент рН).
Количественная оценка возможна при соблюдении постоянства условий эксперимента по следующим признакам: 1) прямое определение по пластинке по площади образовавшегося при проявлении пятна (градуировочный график); 2) определение по интенсивности окраски проявленного пятна (денсиметрический метод, градуировочный график); 3) снимают адсорбент с соответствующей зоны, обрабатывают растворителем, центрифугируют и в полученном растворе вещества определяют каким-либо способом.
Выше описано разделение путем восходящего элюирования. Возможны иные варианты элюирования [244]: нисходящее элюирование; горизонтальное элюирование (круговая хроматография [250]) — пробу наносят по кругу на квадратную пластинку с тонким слоем сорбента (на пересечение диагоналей), в центр подают растворитель, вещества разделяются с образованием концентрических зон; многократное элюирование одним и тем же растворителем (после элюирования пластинку высушивают и операцию повторяют со свежей порцией элюента); ступенчатое элюирование — многократное элюирование разными растворителями; центрифужное элюирование — под действием центробежной силы движение потока растворителей ускоряется в 2—3 раза; градиентное элюирование — состав элюента непрерывно изменяется.
Достоинства ТСХ-анализа
• ТСХ/ВЭТСХ пластинки могут быть использованы в любой момент, обычно не надо проводить их предподготовку;
• на одной пластинке достаточно места для нанесения от 20 до 30 проб или растворов сравнения, что позволяет провести их элюирование в одно и то же время;
• для элюирования всех зон достаточно всего нескольких миллилитров подвижной фазы;
• время самого анализа составляет всего несколько минут, что позволяет очень быстро получить результаты, которые можно перенести на ВЭЖХ (т.е. использовать ТСХ как пробный метод для ВЭЖХ).
Тонкослойная хроматография с обращенной фазой. Сорбент предварительно пропитывают раствором соответствующего реагента, анализируемый водный раствор играет роль подвижной фазы [244].
Многофазная хроматография в тонком слое — хроматографирование на пластинке с двумя слоями разных сорбентов [255].
Осадительная тонкослойная хроматография для разделения и определения некоторых ионов. Пример — разделение Cu2+, Fe3 , Ni2+, Co2+ на слое силикагеля, содержащего 14% 8-гидроксихинолина, или разделение I", Вг~, С1~ на слое силикагеля, пропитанном нитратом серебра. Зоны элементов получаются в виде полос, по ширине которых приближенно оценивают содержание соответствующего элемента [244].
Хромато-масс-спектрометрия — сочетание хроматографии и масс-спектрометрии. Применяют для анализа сложных смесей (до 500 компонентов). Исследование масс-спектра во время прохождения хроматографического пика [247, 256].
Масс-спектрометрия основана на разрушении органической молекулы под действием электронного удара и регистрации массы образующихся осколков. Допустим, что через пары вещества проходит поток электронов, энергию которых можно постепенно увеличивать. Если эта энергия достигнет определенного уровня, то при столкновении электрона с молекулой может произойти отрыв от нее электрона с образованием молекулярного иона М+
ABCD+е ABCD+ +2e-
молекулярный ион
Наименьшая энергия бомбардирующих (ионизующих) электронов, при которой возможно образование из данной молекулы иона, называется энергией ионизации вещества). Энергия ионизации является мерой прочности, с какой молекула удерживает наименее сильно связанный с ней электрон, следовательно, чем прочнее связь электронов с молекулой вещества, тем выше энергия ионизации. Как правило, для органических молекул энергия ионизации составляет 9—12 эВ.
Если энергия ионизующих электронов значительно превышает энергию ионизации, образующийся молекулярный ион получает избыточную энергию, которой может оказаться достаточно для разрыва в нем связей. В результате такого разрыва происходит распад молекулярного иона на частицы меньшей массы (фрагменты). Такой процесс называется фрагментацией. В практике масс-спектрометрии используются электроны с энергией 30— 100 эВ, что намного превышает энергию ионизации и обеспечивает фрагментацию молекулярного иона.
Реальная структура ионов, образующихся под электронным ударом, в подавляющем большинстве случаев не установлена. Для изображения строения фрагментов обычно используются стандартные структурные символы органической химии. Несмотря на условность такого изображения и, в основном, эмпирический характер закономерностей фрагментации, измерение массы образующихся осколков и их относительного количества позволяет получить ценную информацию о строении органических соединений.
Для получения масс-спектра пары вещества небольшими количествами с помощью специальной системы напуска вводятся в ионизационную камеру, где поддерживается глубокий вакуум (остаточное давление около 10-6 мм рт. ст.). Молекулы вещества бомбардируются потоком электронов, излучаемых раскаленным катодом. Разность потенциалов между катодом и анодом ускоряет электроны до определенного уровня энергии (например, до 30 ± ±2 эВ). Образующиеся ионы выталкиваются из ионизационной камеры небольшой разностью потенциалов. Получаемый поток ионов ускоряется, фокусируется сильным электрическим полем и попадает в магнитное поле. В результате бомбардировки молекул вещества электронами образуются частицы, имеющие положительный или отрицательный заряд, а также нейтральные частицы. При прохождении потока частиц через магнитное поле нейтральные частицы не изменяют направления, а положительные и отрицательные отклоняются в разные стороны. Величины отклонения ионов пропорциональны заряду и обратно пропорциональны их массе, иными словами, обратно пропорциональны массе, приходящейся на единицу заряда (m/z).
R=SQRT(2*V*m/z H2)
Где V-напряжение, ускоряющее ионы, m – масса иона, z – заряд, Н – напряженность магнитного поля.
В обычной масс-спектрометрии принято регистрировать только частицы, имеющие положительный заряд. Учитывая, что заряд частицы, как правило, равен единице, величина m/z эквивалентна массе иона.
Если на выходе ионов из магнитного поля установить регистрирующее устройство, то частицы, различающиеся значениями m/z, будут давать раздельные сигналы. Интенсивность сигналов будет пропорциональна количеству частиц с данным значением m/z. Анализ ионов по величине m/z обычно производится при изменении напряженности магнитного поля; ионы постепенно фокусируются -в щель коллектора, где регистрирующее устройство производит запись образующихся электрических импульсов. Это масс-анализатор с однородным магнитным полем.
Квадрупольный масс-анализатор представляет собой квадрупольный конденсатор (рис. 6), к парам параллельных стержней к-рого приложены постоянное напряжение V и переменное высокочастотное V0cos(ot (со-частота, t-время); их суммы для каждой пары равны по величине и противоположны по знаку. Ионы, вылетевшие из ионного источника, движутся в камере анализатора вдоль оси z, параллельной продольным осям стержней, по сложным объемным спиралевидным траекториям, совершая поперечные колебания вдоль осей jc и у. При фиксированных значениях частоты и амплитуды переменного напряжения ионы с определенными значениями m/z проходят через квадруполъный конденсатор, у ионов с др. значениями m/z амплитуда поперечных колебаний достигает такой величины, что они ударяются о стержни и разряжаются на них. Развертка масс-спектра производится путем изменения постоянного и переменного напряжений или частоты. Для совр. квадрупольных масс-спектрометров R = 8000. – разрешающая способность. R = m/m

Рис. 6. Схема квадрупольного масс-анализатора: 1 - высокочастотный генератор; 2-генератор постоянного напряжения; 3-генератор развертки; 4 и 5-источник и детектор ионов.
Детекторы (приемники) ионов помещают на выходе прибора. Для детектирования используют электрометрические усилители, позволяющие измерять ионные токи до 10-14 А, электронные умножители и сцинтилляционные детекторы с фотоумножителем, к-рые обеспечивают счет отдельных ионов (ток 10-19 А) и имеют малую постоянную времени, а также фотопластинки, преимущество к-рьгх в возможности регистрации всех ионов масс-спектра и накопление сигнала.
Хроматомасс спектрометрия.
Хроммасс – LCMS – это хроматограф совмещенный с масс-спектрометром. Современный прибор состоит из автосэмплера Gilson 215 – устройства, позволяющего отбирать много проб одну за другой по заданной программе в автономном режиме под управлением компьютера. Хроматографа, Shimatdzu который так же управляется компьютером, имеется возможность программно изменять состав элюента с течением времени. Так же данный хроматограф может быть использован для препаративной очистки веществ. И собственно масс-спектрометра API – 150.
Основная проблема взаимосвязи ЖХ и МС состоит в сложности перевода растворенных и сольватированных молекул образца в молекулярный газ без значительного его разрушения.

На рис. Показана схема устройства APCI – atmospheric pressure chemical ionization. Образец в растворителе распыляется потоком газа (nebulizer gas) Капли растворителя в потоке газа-носителя проходят через испаритель.
И после окончательного испарения в нем попадают в ионизатор. Ионизасия происходит за счет коронарного разряда на игле. (Corona discharge needle). Электроны от растворителяразряжаются на положительно заряженной игле. После этого образуются кластеры образца с растворителем, затем перенос протона с образованием молекулярного иона МН+.
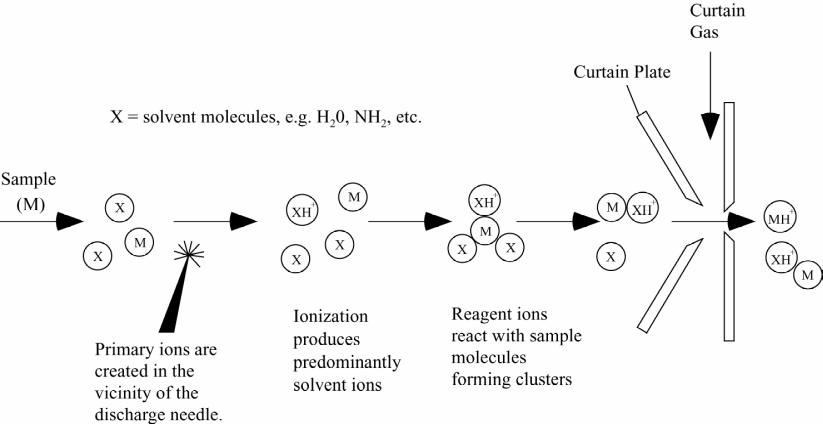
Отрицательный полюс находится на пластине Curtain plate. Ионы ускоряются разностью потенциалов и проходят через диафрагму Orifice в область вакуума, где расположен квадрупольный масс-анализатор. Неионизированные молекулы образца и растворителя отдуваются потоком газа (Curtain gas), который идет между занавесной пластиной и диафрагмой.

Вся система управляется обычным компьютером с помощью программы Analyst 1.3.1. Спектр представляет собой совокупность хроматограмм с использованием в качестве детекторов детектора масс-спектрометра – хроматограмма ионного тока, обычных УФ детекторов на 254 и 215 нм и детектора ELSD – идентификация твердого вещества.
Кроме того имеется возможность получить масс-спектр любого пика в хроматограмме ионного тока, что иногда оказывается очень полезно.
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
4-1. ПРИРОДА СПЕКТРА ПМР
Как и электрон, атомное ядро вращается и характеризуется определенным моментом количества движения ( I ). Момент количества движения строго квантован. Ядро, имеющее нечетное число протонов, при вращении обладает магнитным моментом (), так как любое вращение заряда создает магнитное поле. Величина также квантована, т. е. может принимать только строго определенные значения. В простейшем случае — для ядра водорода (протона)— момент количества движения может иметь значения I = ± ½, магнитный момент также имеет значения = ± ½.
Если поместить вещество в сильное магнитное поле, произойдет определенная ориентация осей вращения содержащихся в нем протонов; эти оси расположатся вдоль направления силовых линий поля. При этом возможны два варианта ориентации, различающиеся энергетическими уровнями: по направлению поля (параллельная ориентация, более выгодная) и против этого направления (антипараллельная ориентация, энергетически менее выгодная).
Если перпендикулярно направлению силовых линий сильного поля приложить относительно небольшое вращающееся магнитное поле и изменять его частоту, то при совпадении частот вращения поля и ядра будет наблюдаться явление резонанса, выражающееся в переориентации осей вращения ядер. Такая переориентация связана с поглощением энергии поля, что легко может быть зарегистрировано. Такое поглощение лежит в основе спектроскопии ядерного магнитного резонанса, или спектроскопии ЯМР.
Наиболее распространенным видом спектроскопии ЯМР является спектроскопия протонного магнитного резонанса, ПМР, основанная на переориентации осей ядер водорода, в котором (в отличие от других перечисленных ядер) резонанс может наблюдаться для распространенного природного изотопа. Кроме того, атомы водорода присутствуют практически во всех органических соединениях.
4-2. МАГНИТНОЕ ЭКРАНИРОВАНИЕ И ХИМИЧЕСКИЙ СДВИГ
Ядра водорода в органических молекулах окружены электронами. Вращение электронов создает свое поле, которое накладывается на внешнее поле, действующее на ядро. Иными словами, электроны заслоняют (экранируют) ядро от внешнего магнитного поля, поэтому напряженность поля в непосредственной близости к ядру отличается от напряженности внешнего магнитного поля. В результате изменения магнитного экранирования изменяется частота вращающегося поля, при которой наблюдается явление резонанса. Это изменение называется химическим сдвигом. Магнитное экранирование и, следовательно, химический сдвиг определяются положением данного протона в молекуле. Для эквивалентных протонов значение химического сдвига одинаково, и они дают один резонансный сигнал. Различающиеся окружением в молекуле протоны обладают различными химическими сдвигами и дают раздельные сигналы, что позволяет определять положение протона в молекуле.
Положение резонансного сигнала зависит от напряженности постоянного внешнего поля (H0), так как эта напряженность определяет силу, ориентирующую ось вращения протона. Для выражения химических сдвигов необходима величина, не зависящая от H0. За международный стандарт принято положение резонансного сигнала тетраметилсилана (CH3)4Si (TMC). Вводимый в раствор вещества эталон должен обладать низкой реакционной способностью, хорошей растворимостью и давать один четкий сигнал в спектре. Кроме того, преимуществом ТМС является положение резонансного сигнала в более сильном поле, чем у подавляющего большинства органических веществ, а также большое количество протонов на единицу массы, что позволяет использовать эталон в минимальных количествах.
Пусть Нэ — напряженность поля, при которой наблюдается резонанс эталона, Н — напряженность поля, при которой наблюдается резонанс данного протона, а H0 — напряженность основного внешнего поля; отношение
(Нэ - Н)/Но
и будет безразмерной величиной, не зависящей от напряженности внешнего поля и характеризующей данный тип протона. Эта величина имеет порядок 10-6, поэтому для значений химического сдвига используют единицы , измеряемые в миллионных долях (м. д.):
![]()
Измерение напряженности поля значительно более сложно и менее точно, чем измерение частоты. В то же время для каждого данного типа протона резонансная частота линейно зависит от напряженности (h — постоянная Планка):
![]()
Поэтому для выражения химических сдвигов используется величина
![]()
где o— рабочая частота; — резонансная частота данного протона; э —резонансная частота протонов эталона.
Получаемый спектр ПМР имеет вид. ТМС сейчас никто не добавляет. Увеличение химического сдвига соответствует переходу в область более слабого поля, т. е. уменьшению степени магнитного экранирования данного протона.
Важное значение имеет также интенсивность сигналов, так как поглощение энергии при данной частоте пропорционально числу протонов, для которых при этой частоте наблюдается явление резонанса. Это позволяет установить, сколько протонов образуют каждый сигнал.
4.3. ПОЛУЧЕНИЕ СПЕКТРОВ ПМР
Для получения спектра ПМР достаточно высокого разрешения используются в основном жидкие маловязкие образцы. Вещество помещают в виде раствора в тонкую (диаметром 5 мм) цилиндрическую ампулу длиной ~ 150 мм. Ампула заполняется на 20—30 мм, для чего требуется около 0,4 мл раствора. Концентрация этого раствора обычно составляет 0,2 моль/л, или 5—20 %, т. е. для приготовления пробы требуется 5—10 мг вещества. Применяемый растворитель в идеальном случае не должен содержать собственных протонов (ССЦ и дейтерированные растворители: D2O, CDC13, C6D6, CD3COCD3 и т. д.). В ампулу вводится также небольшое количество (~1%) ТМС в качестве внутреннего эталона.
Ампула с образцом помещается в мощное постоянное магнитное поле; напряженность поля в зависимости от рабочей частоты составляет 14—117,4 кГс (килогаусс). К однородности этого поля предъявляются очень высокие требования, так как ею в основном определяется качество получаемых спектров. Ампула в приборе вращается, чтобы исключить возможность проявления неоднородности образца. Очень важно отсутствие в образце нерастворимых частиц, которые могут вызвать местное изменение поля.
В перпендикулярном направлении к основному прикладывается вращающееся магнитное поле, имеющее постоянную частоту (например, 100 или 360 МГц или более), и накладывается дополнительное вращающееся поле изменяющейся частоты (например, 0—6000 Гц для рабочей частоты 300 МГц). Поглощение энергии при данной частоте регистрируется специальным датчиком. Спектр представляет собой зависимость интенсивности поглощения энергии от величины , отражающей изменение частоты вращающегося поля. Обычно спектр выглядит как набор узких резонансных сигналов, соответствующих отдельным типам протонов. Для определения интенсивности (точнее, площади) сигналов современные приборы снабжены устройством для электронного интегрирования спектров.
Таким образом, спектр ПМР позволяет определить количество различающихся типов протонов и число протонов каждого данного типа.
Наблюдать протонные спектры на резонансных частотах выше 100 МГц можно только с использованием сверхпроводящих магнитов. Такой магнит имеет следующее устройство: Соленоид, намотанный из сплавов ниобия, погружен в емкость с жидким гелием, которая находится внутри высококачественного криостата (большой цилиндр справа на рис. 1.3). Криостат имеет внешнюю охлаждающую рубашку для охлаждения «радиационного экрана», заполненную жидким азотом. Продуманная конструкция и тщательное изготовление криостата обеспечивают низкий расход жидкого гелия. Его добавляют в криостат каждые 1-5 мес в зависимости от модели конструкции. Внутри отверстия магнита помещен набор градиентных катушек для устранения градиентов поля («шиммы»), а внутри их находится датчик ЯМР (рис. 1.5). Датчик - самая ответственная часть всей системы. Он обеспечивает передачу импульсов к образцу и регистрацию сигналов ЯМР. В спектрометре, MSL-300, датчики вставляются снизу с основания криостата в активную область магнита. При необходимости датчик можно заменить на другой. Образцы для измерений готовятся в обычных цилиндрических ампулах для ЯМР. Образец опускается через верхнее отверстие в зазор криостата и попадает в верхнюю часть датчика. Образец вращается вокруг вертикальной оси с помощью воздушной турбинки. Шиммирующие катушки для настройки однородности поля, датчик и образец находятся при комнатной температуре, хотя совсем рядом с ними поддерживается температура жидкого гелия 4 К.
Пульт спектрометра содержит генератор радиочастотных импульсов и приемник для регистрации сигналов ЯМР. Оба этих блока похожи на обычные радиоустройства. В современных спектрометрах предусматриваются возможности для получения самых разных импульсных последовательностей с различной продолжительностью и фазой, т.е. для осуществления «импульсного программирования». Все функции спектрометра находятся под контролем компьютера, который также используется для обработки данных и представления результатов. Электрические сигналы ЯМР превращаются в цифровые данные для ввода в компьютер с помощью аналого-цифрового преобразователя. Именно он часто является узким местом, ограничивающим класс экспериментов, которые мы можем выполнять.
Все современные ЯМР-спектрометры высокого разрешения работают по принципу импульсного ЯМР. Это означает, что вместо метода непрерывной развертки при котором для увеличения разрешения требуется существенное увеличение времени эксперимента, по данному методу образец облучается коротким и мощным радиочастотным импульсом, а затем фиксируется затухающий отклик, который называют fid – или ССИ – спад свободной индукции. Затем мы можем повторить эксперимент для улучшения отношения сигнал/шум. При увеличении количества повторений в н раз соотношение улучшается в корень из н раз. После выполнения достаточного числа повторений мы получаем в свое распоряжение данные, содержащие информацию обо всех частотах в спектре ЯМР, однако в непривычной для нас форме. Это временное представление спектра. Для перехода к привычной форме или к частотному представлению спектра необходимо провести Фурье-преобразование. ФИД фиксируется время порядка 1с. У нас 1.35с.
Время накопления AQ = 1.35 определяет разрешение спектра. А частота с которой выбираются точки в ФИДе при его оцифровке определяют ширину спектра. До преобразования с ФИДом можно сделать необходимые операции, например экспоненциальное умножение для улучшения соотношения сигнал-шум.


Еще одно преимущество импульсной ЯМР-спектроскопии заключается в том, что мы можем давать не один импульс, а целые последовательности импульсов через заданные интервалы. Такие эксперименты дают интересные результаты. В частности, на этом принципе основано получение двумерных спектров.
Шимовые катушки служат для настройки однородности магнитного поля в спектрометре. В принципе шиммы нужно настраивать по ФИДу – стараться получить как можно более длинный фид след. Можно взять больше AQ. И добиться лучшего разрешения. Именно поэтому настройку шиммов часто называют настройкой разрешения. Однако на практике шиммы настраивают по сигналу дейтерия в растворителе. Амплитуда сигнала лока зависит от нескольких факторов и в том числе от однородности поля. Поэтому ее можно использовать в качестве критерия при шиммировании. Изменением тока, проходящего через соответствующие катушки добиваются максимальной амплитуды сигнала лока. При первоначальной настройке настраиваются все шиммы. Затем, при переходе от образца к образцу только Z и Z2.
На магнитное экранирование протона влияет множество факторов, в связи с чем зависимость между положением ядра в молекуле и соответствующим химическим сдвигом носит эмпирический характер. Основным из таких факторов является электронная плотность вокруг данного протона. Чем выше эта плотность, тем больше ее влияние на внешнее поле и, следовательно, тем в более сильном поле проявится резонансный сигнал. Влияние этого фактора соответствует тому, что протон, обладающий более кислыми свойствами (с меньшей плотностью электронной оболочки) резонирует в более слабом поле. На электронную плотность вблизи ядра существенно влияет индукционный эффект заместителя и присутствие соседних непредельных группировок, так как в последнем случае благодаря эффекту сопряжения электроны связи С—Н смещаются на соседнюю связь С—С. Таким образом, по величине химического сдвига может быть определено положение протона в органической молекуле. Однако сигналы от протонов, незначительно отличающиеся по химическому сдвигу, могут перекрываться полностью или частично. Так, незначительно различающиеся по положению метиленовые группы в ациклических цепях или циклических системах часто сливаются в один широкий сложный сигнал.
СПИН-СПИНОВЫЕ ВЗАИМОДЕЙСТВИЯ
Спин протона практически равновероятно имеет значения +½ и —½ На магнитное экранирование каждого данного протона оказывает влияние спин соседнего протона, который может быть различен и поэтому дает два различающихся поля: одно увеличенное, другое — уменьшенное. С удалением ядер друг от друга эффект резко падает. Влияние на магнитное экранирование протона спина другого неэквивалентного протона, расположенного при соседнем углеродном атоме, называется спин-спиновым взаимодействием. Это явление приводит к усложнению спектра.
Если протон при соседнем углеродном атоме отсутствует (например, в группировках —ОСН3, —СОСН3), спин-спиновое взаимодействие не проявляется; в спектре возникает одиночный сигнал или синглет. При наличии «соседних» протонов наблюдается расщепление сигналов, характер которого зависит от числа взаимодействующих ядер. Рассмотрим простейшие спин-спиновые системы. С целью упрощения написания спины -+ ½ и — ½ обозначим соответственно как А и Б.
Система -СН—СН- реализуется, например, в соединении
O=CH1-CH2Cl2
Рядом с протоном Н1 находится протон Н2, имеющий равновероятно спин А или Б (Н2A и Н2Б ). Следовательно, половина протонов Н1 будет иметь одно магнитное экранирование, тогда как другая половина — другое. Соответственно резонанс для одной половины протонов Н1, находящейся под воздействием Н2 со спином А, пройдет при иной частоте поля, нежели резонанс у другой половины, на которую воздействует Н2Б Следовательно, сигнал от протона Н1 расщепится на два компонента равной интенсивности, т. е. превратится в дублет. Аналогично, расщепление в дублет будет наблюдаться для Н2, рядом с которым равновероятно могут находиться протон На и протон Нб- Спектр системы будет таким (нарисовать спектр аль – 9 хлор - 6). Расстояние между компонентами того и другого дублета одинаково, так как расщепление обусловлено одной причиной. Это расстояние, выраженное в герцах, называется константой спин-спинового взаимодействия (обозначается буквой J).
Система
СН—СН2 — существует, например, в соединении
(С12СН—СН2С1)
С12СH1—СH22—С1
Каждый из эквивалентных протонов Н2 метиленовой группы находится в соседнем положении с протоном Н1, который равновероятно имеет спин А и Б. По аналогии с описанным выше сигнал от протонов метиленовой группы (Н2) будет представлять собой дублет. Поскольку протоны Н2 также равновероятно имеют спин А или Б, для протона Н1 возможны следующие комбинации спинов соседних протонов Н2:
АА АБ ББ
БА
Комбинации АБ и БА эквивалентны по образуемому магнитному полю. Следовательно, сигнал от протона Н1 расщепится на три компонента с соотношением интенсивностей 1:2:1, т. е. будет иметь форму триплета. Спектр ПМР для данной системы будет иметь вид:

Система СН—СН3 реализуется, например, в соединении
С12 H1 С-СН32
По аналогии с описанным выше, сигнал от трех протонов метильной группы (Н2) будет представлять собой дублет. Для протона Н1 возможны следующие комбинации спинов соседних протонов Н2
ААА ААБ АББ БББ
АБА БАБ
БАА ББА
Следовательно, сигнал протона Н1 расщепится на четыре компонента с соотношением интенсивностей 1:3:3:1, т. е. будет представлять собой квартет.
Система —СН2—СН3 присутствует, например, в этилбромиде
Спектр будет иметь вид.
Аналогичным образом может быть рассчитана форма сигналов в более сложных спин-спиновых системах. В общем виде, если данный протон взаимодействует с n эквивалентными протонами, его резонансный сигнал должен состоять из n + 1 компонент. Соотношение интенсивностей отдельных линий отвечает статистическому вкладу данной комбинации спинов.
Таким образом, в простейших случаях по мультиплетности сигнала можно определить число протонов при соседних углеродных атомах, или, иными словами, группы, соседние по отношению к данной связи С—Н. Следовательно, спин-спиновое взаимодействие дает дополнительную ценную информацию о строении исследуемого вещества.
Если в системе наблюдается большое количество спин-спиновых взаимодействий, особенно между протонами с близким характером магнитного экранирования, сигнал становится многокомпонентным, иногда неправильной формы. Такие сигналы довольно распространены и носят название сложных мультиплетов.
Значения констант спин-спинового взаимодействия варьируют в широких пределах —от 1 до 20 Гц, в зависимости от магнитных свойств взаимодействующих ядер и их взаимного расположения в пространстве. Для каждого типа спин-спиновой системы величина J примерно постоянна и не зависит от напряженности внешнего поля, поскольку определяется свойствами самих ядер. Например, для следующих систем J имеет значения (Гц):

В сложных спектрах путем сравнения величин J для различных сигналов удается установить, какие из сигналов образованы соседними (взаимодействующими) протонами, так как у этих сигналов константы спин-спинового взаимодействия будут одинаковы.
Таким образом, спектр ПМР дает нам пять основных аналитических критериев: общее число сигналов (число типов неэквивалентных протонов); интенсивность сигналов (число протонов каждого данного типа); химический сдвиг (положение протона в молекуле); мультиплетность, или структура, сигнала (число протонов при соседних углеродных атомах); константы спин-спинового взаимодействия (особенности расположения протонов в пространстве). Указанные критерии позволяют получить ценные сведения о строении вещества.
Спектроскопия ЯМР является наиболее информативным из всех используемых в настоящее время физико-химических методов исследования органических веществ. Поэтому, несмотря на относительно сложную конструкцию радиоспектрометров, данный метод находит широкое применение в современной лабораторной практике.
Разобрать пару спектров.
Разобрать двумерные спектры
4. Современные синтетические методы.
4.1. Реакции получения гетероциклов.
Как отмечалось ранее, в качестве базовой структуры для получения биологически активных соединений наиболее перспективны гетероциклы.
Для проведения комбинаторной стадии т.е. для возможности синтеза библиотеки структурных аналогов в молекуле билдинг-блока необходимо наличие активной функциональной группы – такой как:
![]()
Таким образом молекула билдинг-блока в общем виде должна выглядеть следующим образом:
![]()
Возможны следующие варианты конструирования билдинг-блоков такого строения:
-
Введение активной функциональной группы в уже имеющийся коммерчески доступный гетероцикл, например:

-
Синтез гетероцикла с уже имеющейся активной функциональной группой, например:

-
Синтез гетероцикла с последующей его функционализацией, например:

-
Дальнейшая функционализация уже имеющихся билдинг-блоков с целью расширения гаммы синтезируемых соединений – т.н. реакции удлинения:

Для функционализации широко используется реакция сх.
О реакции , методах выделения и очистки.
Так же можно использовать реакцию нитрования
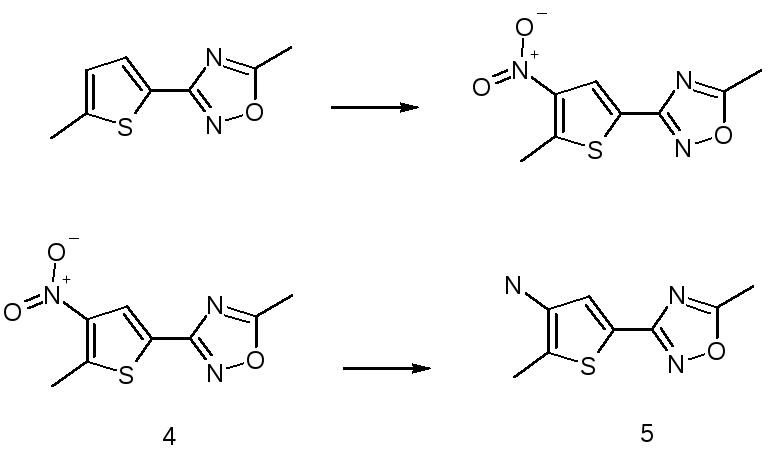
Так же элек. Замещ.
Обычно используют или 100% ак, или смесь 100% ак и ск или используют нитрат К или На в конц. Ск. В данном случае реакция идет при комн. Т.
Выделение в лед.
Для восстановления используют хлористое олово, соотношение нитра:олово:солянка = 1:4:8 – в спирте. гидразин на никеле, водород под давлением на никеле или палладии.
Про достоинства и недостатки каждого метода.
Про удлинения – реакция Шоттена-Баумана, пикотаты с последующим гидролизом , про пиперазин.
СИНТЕЗ СУЛЬФОПРОИЗВОДНЫХ НА ОСНОВЕ 3Н-БЕНЗОКСАЗОЛ-2-ОНА
По каждому циклу – что за реакция, условия получения и выделения, доказательство структуры.
Синтез 3Н-бензоксазол-2-она 5 осуществлялся взаимодействием 2-аминофенола 4 с мочевиной в присутствии соляной кислоты при нагревании (схема1) [5].

Спросить, что за реакция – нуклеофильное замещение.
Реакция идет с отщеплением аммиака, кот. Связывается HCl.
12 часов при кипячении соотношение аф:моч: HCl=1:1.2:4
Найти спектр
Сх б-на
Реакция электрофильного замещения ориентанты первого и второго рода, куда пойдет сх?
Система активирована, поэтому условия сх мягкие.
Р-я сх идет в два этапа
Необходимо отделить от воды – экстракция. – Позволяет отделить от воды и от сульфокислоты. Очистка – флэш-хроматография. Возможна перекристаллизация. Сущность того и другого.
2-оксо-2,3-дигидро-бензоксазол-6-сульфонилхлорид (6): К 40 мл хлорсульфоновой кислоты (0.5 моль), охлажденной до 10 С, при интенсивном перемешивании порциями добавляли 13.5 г (0.1 моль) 5 в течение 30 минут. Реакционную смесь перемешивали при 20 С в течение 20 минут, затем нагревали до 60 С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 300 мл диэтилового эфира. Органический слой промывали водой и сушили над хлористым кальцием, растворитель отгоняли. Продукт перекристаллизовывали из толуола. Выход 6 – 10.5 г (45%), т. пл 188-189 С.
Модификация б-на
Метилирование: р-я нуклеофильного замещения
Депрототнирование происходит легко.
Реакцию проводят в ДМФА в присутствии поташа. Т-
Дальше – сульфохлорирование:
3-Метил-2-оксо-2,3-дигидро-1,3-бензоксазол-6-сульфонилхлорид (10): К 20 мл хлорсульфоновой кислоты (0.3 моль), охлажденной до 30-40 С, при интенсивном перемешивании порциями в течение 30 минут добавляли 14.9 г (0.1 моль) 9. Реакционную смесь перемешивали при 40 С в течение 20 минут. Затем добавляли 25 г PCl5 (0.12 моль) при 50-70 С в течение 1 ч. Реакционную массу нагревали до 80 С и перемешивали в течение 1 ч. После охлаждения реакционную массу выливали на 500 г измельченного льда, продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Экстракт пропускали через силикагель, отгоняли хлороформ под вакуумом, и сушили. Перекристаллизовывали из толуола. Выход соединения 10 – 7.5 г (51%), т. пл. 159-160 С.
Добавлением PCl5 можно добиться снижения расхода ХСК, смягчения условий на стадии хл-я.
Доказательство положения сульфохлорирования проведено на примере:

Рис. 3. Структура соединения 11е.

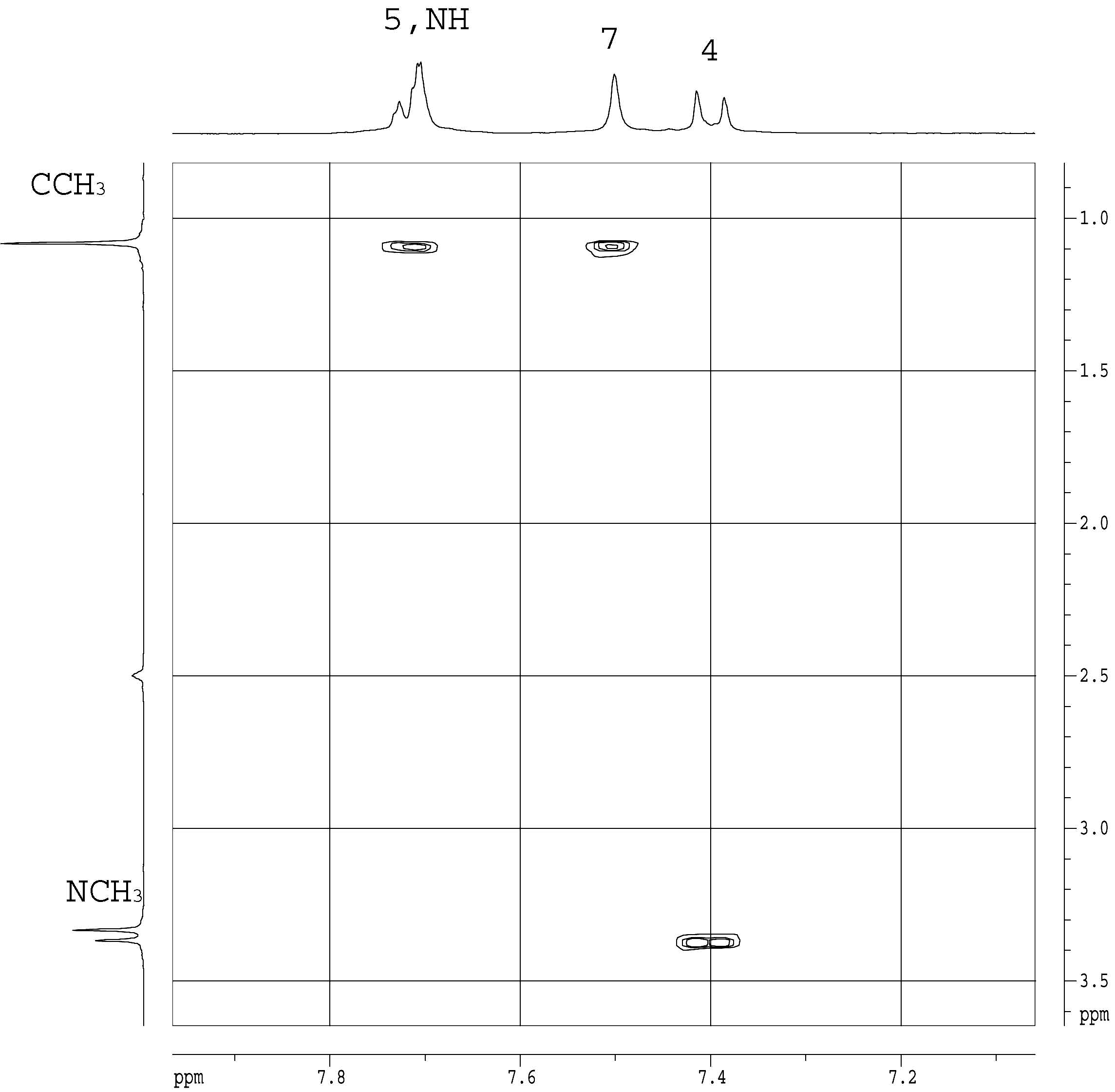
Рис. 4. Двумерный корреляционный спектр 1Н-1H NOESY соединения 11е.
В 1Н-1Н NOESY спектре соединения 11е наблюдается кросс-пик, характеризующий взаимодействие протона группы NCH3 (хим. сдвиг 3,4 м.д.) с одним ароматическим протоном (протон при 4С, 7,4 м.д.). Сигнал этого протона представляет собой дублет с константой J=8,3 Гц, характеризующей его орто-взаимодействие с еще одним протоном (протон при 5С, 7,7 м.д.). Последнее было бы невозможным в случае местонахождения сульфогруппы в положении 5-. Одновременно в пектре NOESY наблюдается кросс-пик, характеризующий взаимодействие этих двух протонов. Кроме того, на спектре присутствует кросс-пик, характеризующий взаимодействие протонов при 5С и 7С (7,5 м.д.) с протонами CH3 групп аминного фрагмента (1’С, 1,05 м.д.), причем сигнал протона при 7С представляет собой синглет.
Алкилирование б-на эфиром хлоруксусной кислоты. Так же нуклеофильное замещение, поташ, 80 гр, 2 часа. Очистка – крист.
Сульфохлорирование:
Этиловый эфир (6-хлорсульфонил-2-оксо-бензоксазол-3-ил)-уксусной кислоты (15): К 20 мл (0.3 моль) хлорсульфоновой кислоты, охлажденной до 10 С, при интенсивном перемешивании порциями добавляли 23.3 г (0.1 моль) 14 в течение 30 минут. Реакционную смесь перемешивали при 30 С в течение 30 минут. Затем добавляли 25 г (0.12 моль) PCl5 в течение 1 ч постепенно повышая температуру с 30 до 50 С. Затем реакционную смесь нагревали до 60 С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Органический слой промывали водой, сушили над хлористым кальцием, затем растворитель отгоняли под вакуумом. Продукт перекристаллизовывали из толуола. Выход соединения 15 – 15.4 г (48%), т. пл. 136-137 С.
Дальше реакция ацилирования амина сульфохлоридом.
Дальше – гидролиз эфира. Возможны два типа гидролиза сложных эфиров – щелочной и кислотный. Щелочной рушит цикл. В кислоте цикл устойчив.
Общая методика получения кислот 12, 12’, 17, 17’: К раствору, содержащему 0.1 моль эфира кислоты (11а, 11б, 16, 16’ соответственно) в 200 мл диоксана добавляли при перемешивании 200 мл соляной кислоты (36%). Реакционную смесь нагревали до 100 С и перемешивали в течение 2 ч. Затем охлаждали до комнатной температуры. Продукт отфильтровывали, промывали водой и перекристаллизовывали из изопропилового спирта.
Этиловый эфир (6-хлорсульфонил-2-оксо-бензоксазол-3-ил)-уксусной кислоты (15): К 20 мл (0.3 моль) хлорсульфоновой кислоты, охлажденной до 10 С, при интенсивном перемешивании порциями добавляли 23.3 г (0.1 моль) 14 в течение 30 минут. Реакционную смесь перемешивали при 30 С в течение 30 минут. Затем добавляли 25 г (0.12 моль) PCl5 в течение 1 ч постепенно повышая температуру с 30 до 50 С. Затем реакционную смесь нагревали до 60 С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Органический слой промывали водой, сушили над хлористым кальцием, затем растворитель отгоняли под вакуумом. Продукт перекристаллизовывали из толуола. Выход соединения 15 – 15.4 г (48%), т. пл. 136-137 С.

 С
С
Дальше – удлинения с бок-пиперазином. Так же кислотный гидролиз.