

Yuriy Kruglyak. Quantum Chemistry_Kiev_1963-1991
.pdfwhich is a Coulomb-type term connecting the states with impulses k and k +π (these states are already connected in the second term in Eqs (265) and (267), thus we continue to keep the terms of this kind); and finally
(iv) k1 = k4 +π , namely,
|
γ12 |
|
∑ |
|
ˆ+ |
ˆ |
|
ˆ+ |
ˆ |
|
|
|
− |
N k |
Bk |
+π,σ Bk |
,σ′Bk |
+π,σ′Bk |
,σ cos(k1 |
−k2 ) , |
(273) |
||||
|
,k ,σ |
,σ′ |
1 |
|
1 |
2 |
2 |
|
|
|
||
|
1 |
3 |
|
|
|
|
|
|
|
|
|
|
which is an exchange-type |
term, |
connecting the k |
and k +π |
states. Writing |
||||||||
cos(k1 − k2 ) as
sin k1 sin k2 + cosk1 cosk2 , and reffering to subsequent integration, one can reach further
simplification of the Hamiltonian due to the fact that the ground-state everage of some terms appearing vanish, thus,
ˆ+ ˆ |
≈δσσ′, |
ˆ+ ˆ |
ˆ+ ˆ |
Bkσ Bkσ′ |
Bkσ Bkσ sink = 0, |
Bkσ Bk+π,σ cosk = 0 . |
Introducing two new operators
ˆ |
1 |
|
ˆ+ |
ˆ |
ξσ = |
N |
|
∑Bkσ Bk+π,σ sink, |
|
|
|
k |
(274) |
|
|
1 |
|
|
|
|
|
ˆ+ |
ˆ |
|
ηˆσ = |
N |
|
∑Bkσ Bkσ cosk, |
|
|
|
k |
|
|
one ia able to rewrite the reduced Hamiltonian in the form
ˆ |
|
|
∑ |
|
|
|
|
∑ |
ˆ |
U −2γ12 |
∑ |
ˆ |
|
ˆ |
|
|
|
∑( |
|
2 |
ˆ |
2 |
ˆ2 |
H |
= |
2 |
σ |
+ |
2 |
∆β |
σ |
|
|
σ |
|
−σ |
−γ |
12 |
σ |
|
σ σ ) . (275) |
||||||
|
|
β ηˆ |
|
|
ξ + |
2 |
|
∆ ∆ |
|
|
ηˆ |
|
+ ∆ +ξ |
||||||||||
|
|
|
σ |
|
|
|
|
σ |
|
σ |
|
|
|
|
|
|
σ |
|
|
|
|
|
|
This reduced Hamiltonian is formally very similar to the reduced Hamiltonian solved in the Bardeen – Cooper – Schrieffer (BCS) theory of superconductivity. As has been proved by Bogolyubov [120, 121], its SCF solution for a large system ( N → ∞ ) asymptotically coinsides with the exact one. Thus, what we have to do now is to solve the wave equation with Hamiltonian (275) using the SCF method. It seems to be convenient in our case to write the wave equation in the form of equation of motion.
Let us use the standard Bogolyubov transformation
ˆ |
ˆ |
ˆ |
(276) |
bkσ =Ukσ Bkσ +Vkσ Bk+π,σ |
|||
to define the new operators bˆk+σ ,bˆkσ , satisfying the equation of motion
ˆ |
ˆ |
ˆ |
(277) |
[bkσ ,H ] = λkσbkσ . |
|||
520
If the coefficients Ukσ ,Vkσ in |
(276) are found to satisfy Eq. (277), then the |
|||||
transformation (277) diaginalize Hamiltonian (275). |
|
|||||
Requiring the new operators (276) to be of the Fermi-type |
|
|||||
ˆ+ |
ˆ |
|
|
|
δσσ′ , |
(278) |
[bkσ |
,bk′σ′]+ =δkk′ |
|||||
one obtains the following relation for Ukσ ,Vkσ : |
|
|
|
|||
|U |
kσ |
|2 |
+|V |
|2 |
=1. |
(279) |
|
|
kσ |
|
|
|
|
Substituting Eqs (276) and (275) into (277), and performing the calculations required using (278), one obtains the system of two nonlinear equations with respect to Ukσ ,Vkσ . Linearizing these equations, which corresponds to the SCF procedure, and
using (279), one obtains the solution in the form
|
|
|
λ |
|
|
|
|
|
|
2 |
cos |
2 |
|
2 |
sin |
2 |
k +U |
2 |
2 1/2 |
, |
(280) |
|||||||||||||||
|
|
|
|
= ±(4β |
|
|
|
k +4∆β |
|
|
|
|
∆−σ |
) |
|
|
||||||||||||||||||||
|
|
|
|
|
kσ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
β |
cos k |
|
|
|
|
|
|
|
|
|
|
|||||||
|Ukσ | |
= |
2 |
± |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
|
|||||||||||
|
|
2 |
cos |
2 |
|
2 |
sin |
2 |
k +U |
2 |
2 |
1/2 |
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
(4β |
|
|
k +4∆β |
|
|
|
|
∆−σ |
) |
|
|
, |
(281) |
|||||||||||||
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
β |
cos k |
|
|
|
|
|
|
|
|
|||||||||
|V |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
| |
= |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
|
||||||
|
|
|
2 |
|
|
|
2 |
|
2 |
|
|
|
2 |
|
|
|
2 |
2 |
1/2 |
|
||||||||||||||||
kσ |
|
|
|
|
|
|
|
|
cos |
|
sin |
k +U |
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
(4β |
|
|
k +4∆β |
|
|
|
|
∆−σ |
) |
|
|
|
|
|||||||||||||
where |
|
|
π |
|
|
|
|
|
|
π |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
− |
< k < |
, |
|
|
= |
β −γ12 |
ηˆσ |
, |
|
|
|
|
= ∆β −γ12 |
ξσ . |
(282) |
||||||||||||||||||||
|
2 |
2 |
|
β |
|
|
|
∆β |
||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
To make the solution complete, we have to calculate  ∆σ
∆σ  ,
,  ηˆσ
ηˆσ  , and
, and  ξσ
ξσ  , where
, where
the everaging is implied over the just found ground state, corresponding to all states λkσ occupied with the minus sign in Eq. (280), i.e. the ground state has the form
|
|
|
|
N |
(−) ˆ+ |
|
|
|
|
(283) |
|
|
|
|
|
|
|
||||
|
|
|
|
ψ0 = ∏ |
bki |
|
0 , |
|
|
|
|
|
|
|
i=1 |
|
|
|
|
|
|
where the operators |
(−) ˆ+ |
are defined by Eqs (276) and (281) with the lower sign in |
||||||||
bk |
||||||||||
(281). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ˆ |
|
ˆ |
To perform the required calculations, one should express Bk σ and |
Bk +π ,σ in terms |
|||||||||
of the operators |
(−) ˆ+ |
and |
(+) ˆ+ |
|
|
|
|
|
|
|
bk |
bk ; and after substituting them into (274) to average ∆σ , |
|||||||||
ηˆσ , and |
ξσ over the ground state (283). Taking into |
account that |
only terms like |
|||||||
(−) ˆ |
(+) ˆ+ |
|
|
|
|
|
|
|
|
|
bk and |
bk contribute to the ground state average values, one obtains the following |
|||||||||
system of coupled integral equations with respect to ∆σ |
, ηˆσ , and |
ξσ |
: |
|||||||
521
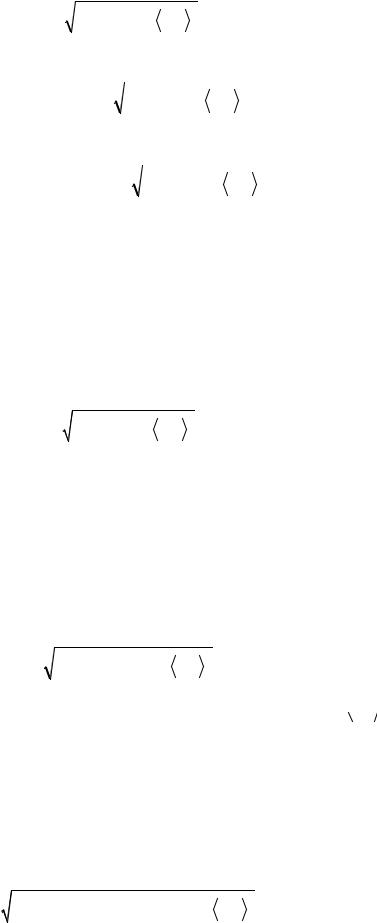
|
|
Uπ |
π∫/2 |
|
|
|
|
|
|
|
|
|
|
dk |
|
|
|
|
|
|
|
|
|
|
|
=1, |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
ε |
2 |
(k) +U |
2 |
|
∆σ |
2 |
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
2γ |
|
|
|
π /2 |
|
|
|
|
|
|
|
|
|
cos2 k dk |
|
|
−1 |
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
β = β 1 |
− |
|
|
12 |
|
∫ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
, |
(284) |
|||||||||||
|
|
π |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
ε |
2 |
(k) +U |
2 |
|
|
2 |
||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
∆σ |
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2γ |
|
π /2 |
|
|
|
|
|
|
sin2 k dk |
|
|
|
−1 |
|
|
||||||||||||||||||
∆β = ∆β |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
1 |
+ |
|
|
12 |
|
|
∫ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
|
||||||||||
|
|
π |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
ε |
2 |
(k) +U |
2 |
|
∆σ |
2 |
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
where |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
ε |
2 |
|
|
|
|
|
|
2 |
cos |
2 |
|
|
|
|
|
|
|
|
2 |
sin |
2 |
k). |
|
|
|
|
|
||||||||||||||
|
(k) = 4(β |
|
|
|
|
|
k + ∆β |
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
This system of equations may be solved iteratively, but in |
the |
usually assumed |
|||||||||||||||||||||||||||||||||||||||
case of γ12 β,U it breaks into |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
∆β ≈ ∆β, |
|
|
β ≈ β, |
|
|
|
|
|
|
|
|
||||||||||||||||||||||||
and |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Uπ π∫/2 |
|
|
|
|
|
|
|
|
|
|
dk |
|
|
|
|
|
|
|
|
|
=1, |
|
|
|
|
|
(285) |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
ε |
2 |
(k) +U |
2 |
|
∆σ |
|
2 |
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
where |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ε2(k) = β |
2 |
|
+ β |
2 |
+2β β |
2 |
cos2k, |
|
|
|
|
|
|||||||||||||||||||||||||||||
|
|
|
|
|
1 |
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
β = β + 1 ∆β, |
|
|
|
|
β |
2 |
|
= β − 1 ∆β, |
|
|
|
|
|
||||||||||||||||||||||||||||
|
|
1 |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
the last equation being the gap equation that, in the case of the regular chain structure (no bond alternation), transforms to equation for the correlation gap [17, 111]
Uπ |
π∫/2 |
|
|
|
|
dk |
|
|
|
=1. |
(286) |
|
|
|
|
|
|
|
|
||||
|
2 |
|
2 |
2 |
2 |
||||||
|
0 |
|
4β |
|
cos |
k +U |
|
∆σ |
|
||
As has been stated in [187], Eq. (286) has a nonzero solution  ∆σ
∆σ  ≠ 0 for all values of the parameters, which we will denote as ∆0 .
≠ 0 for all values of the parameters, which we will denote as ∆0 .
Let us now return to the general case of Eq. (285), assuming that
β(δ x) = β0e−αδ x ,
where δ x denotes displacement from the regular, equal-bond configuration. Thus, Eq. (285) may be rewritten as
U π /2 |
|
|
dk |
|
|
|
|
|
|
|||
π |
∫ |
|
|
|
|
|
|
|
|
|
=1. |
(287) |
|
|
|
|
|
|
|
|
|
||||
(β1 − β2 ) |
2 |
2 |
2 |
k +U |
2 |
2 |
||||||
|
0 |
|
|
+4β0 |
cos |
|
∆σ |
|
||||
522
Comparing (287) with (286) one easily concludes that if ∆0 is the solution of (286) then the solution of (287) is given by
2U 2  ∆σ
∆σ  2 +(β1 − β2 )2 =U 2
2 +(β1 − β2 )2 =U 2  ∆σ
∆σ  02 ,
02 ,
so that
∆σ |
2 = ∆σ |
2 |
− |
1 |
|
β1 |
− β2 |
2 . |
(288) |
|
|
0 |
|
2 |
|
|
U |
|
|
We are now in a position to turn to the final step of the treatment, namely, calculation of the total energy and minimization of it with respect to δ x .
Substituting Eqs (275) and (280) – (283) into the usual expression for the total electronic energy in the ground state
Eel =  ψ0 Hˆ ψ0
ψ0 Hˆ ψ0  ,
,
one obtains
|
|
el = U |
+∑λk |
+U∆2 . |
(289) |
|
E |
||||||
4 |
k |
|
|
|
||
It is important to recognize that according to (280) and (288) |
|
|||||
|
|
λ (β − β |
2 |
)2 /U |
|
|
|
|
k |
1 |
|
|
|
is independent on δ x because the only term containing this dependence cancels in the
expression for λk . Thus, assuming δ x |
to be small, which results in |
β1 − β2 ≈ δ x , one |
|||
can rewrite (289) in the form |
|
|
|
||
|
|
= E − E (δ x)2 . |
(290) |
||
E |
|||||
|
el |
0 |
1 |
|
|
Adding the core deformation energy |
1 к(δ x)2 , |
|
|||
|
E |
= |
|
||
|
|
core |
|
2 |
|
one obtains the total energy of the chain in the form |
|
||||
E = E +α(δ x)2 . |
(291) |
||||
0 |
|
|
|
||
This expression, when minimized with respect to δ x , gives us a solution δ x = 0 only in the case
α = E1 − 12 к > 0,
which obviously corresponds to a vanishing bond correlation and to the energy gap (286) of the pure “correlation” type.
523

In the opposite case of α < 0, the total energy does not exibit a minimum at all,
decreasing formally |
to − ∞ when |
δ x |
increases. Nevertheless, taking into account |
(288), one can see |
that for some |
δ x |
and corresponding β1 − β2 , value of ∆2 |
becomes negative, which evidently means that our solution fails completely. Here we should remember that apart from the solution described by Eqs. (284) and (286), which is the non-trivial solution of the UHF SCF equations, we always have the trivial solution ∆ = 0 , corresponding to the usual HF SCF procedure. We have used the non-trivial solution in the case of the regular chain structure (no alternation) because in this case it corresponds to a lower energy than the trivial solution [174]. However, for α < 0 this non-trivial solution does not minimize the total energy, and for some δ x in the process of its increasing we get ∆ = 0 ; at this point we should jump to the trivial solution because the non-trivial one ceases to exist. Hence, in this case we have an alternating-bond chain with a vanishing ( ∆ = 0 ) contribution of the Mott-type correlation to the creation of the energy gap; while the gap due to the bond alternation should be calculated in a quite different way [171 – 173]. The results is well known: in the absence of the Mott-type contribution, the Peierls-type transition to a semiconducting state necessarily takes place, and the gap obtained can be approximately calculated as
∆alt =8β0e−к/2β . |
(292) |
Therefore, at least at zero temperature the picture is clear: depending on the numerical values of the parameters involved the quasi-one-dimensional chain represents either a Mott-type or a Peierls-type semiconductor, but not their combination, and the choice should be done by comparison of the total energies of both states. Roughly speaking, it may be stated that the real state is the state with the larger gap calculated neglecting the possibility of the other state available. In fact, in addition to the criterion mentioned above for the Mott metal – dielectric transition, one more criterion should be formulated determining the value of the gap arised due to Peierls instability. However, it is evident that if we have U | β | , then the correlation gap is large, ca. U, very likely larger than the gap due to the lattice distortion, and the situation is reversed for U < | β |.
8.2. Finite Temperatures
Let us now consider the same question of the possible combined nature of the energy gap in the case of a finite temperature. Only the general method of calculations and the final results will be presented below. For details of calculations see [172].
524
To get the temperature dependence of all the values we are interested in the following procedure may be proposed: in all equations used the average over the ground state should be replaced by statistical average calculated as
|
ˆ |
ˆ |
ˆ |
|
ˆ |
|
|
|
−H /kT |
/ Spe |
−H /kT |
. |
(293) |
||
A = SpAe |
|
|
|||||
Bearing in mind that the Hamiltonian expressed in terms of the operators bˆk+ ,bˆk at (276) – (282) is diagonal, standard equation (293) is reduced to
|
|
|
ˆ |
|
|
|
|
|
ˆ |
klσ |
, |
(294) |
|||
|
|
|
|||||
A = ∑ηk l,σ (T ) klσ |
A |
|
|||||
|
|
k,l |
|
|
|
|
|
where ηk l is the average number of particles in the state (k,l) with k stands for the
quasi-impulse and l – for the zone number. During all transformations the Fermi character of quasi-particles has been required (see Eq. 278), hence
η |
(T ) ={exp[ε |
k l,σ |
/ kT ]+1}−1, ε |
k l,σ |
≡ λ |
. |
(295) |
k l,σ |
|
|
kσ |
|
|
For the temperature dependent energy gap playing a central role in all the treatment using Eqs (278) and (294) one obtains
|
|
1 |
ˆ+ |
ˆ |
|
|
∆σ = |
N |
∑kl ηkl Bk+π,σ Bkσ . |
(296) |
|||
Substituting Eqs (295) and (276) – (280) into (296) and performing the |
||||||
calculations required which are very similar to those leading to |
(285) one obtains |
|||||
the following equation determining |
|
|
||||
∆σ (T ) , namely: |
|
|
|
|
||
|
U |
π |
th E(k) /2kT dk =1, |
(297) |
||
|
2π |
∫ |
||||
|
E(k) |
|
|
|||
|
|
|
0 |
|
|
|
where
E(k) =[(U∆)2 +ε2(k)]1/2, |
|
|
ε2(k) = |
(β − β |
2 |
)2 +4β2 cos2k . |
(298) |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
0 |
|
|
Introducing the density of states, one can transform (297) into the form |
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
U |
ε2 +∆2 |
|
|
|
thε /2kT |
|
|
|
|
|
|
|
|
|
|||||||
π |
∫ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
dε =1, |
|
(299) |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
ε |
2 |
+ ∆ |
2 |
|
2 |
+ ∆ |
2 |
−ε |
2 |
|
|||||||||||
∆ |
|
|
|
εF |
|
|
|
|
|
|
|
|
|||||||||
where the same assumption β(δ x) ≈ β0e−δ x |
and notation εF |
= | β0 | have been used, but |
|||||||||||||||||||
now |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆2 =| β − β |
2 |
|2 + ∆σ |
2 . |
|
|
(300) |
|||||||||||
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
||
525

Equation (299) may be considered in the same way as it has been done concerning Eq. (287). The one-electron energy levels which are now temperature dependent are also independent on δ x and all the discussion following Eq. (291) may be repeated leading to the same conclusions at the finite temperature as were arrived at in the case of zero temperatute.
Let us |
now |
note that if our system is a Mott-type semiconductor with |
(β1 − β2 ) = 0 |
and |
∆σ ≠ 0, then Eq. (299) becomes similar to the Peierls gap in the |
chain with bond alternation, namely, both of them are BCS-type gaps in superconductors. Unfortunately this does not provide us much information on the nature of metal – Mott semiconductor phase transition because low-lying triplet and singlet excitations should be taken into account before one treats the quasi-ionic states with higher energies; but only these later states may be considered using the standard UHF procedure.
9. Coexistence of Mott and Peierls Instabilities in Quasi-one-dimensional Systems
The quasi-one-dimensional conductors have so far being studied are of interest for both theoreticiants and experimentators. This interest, on the one hand, is due to advances in synthesis of polyacetylene (PA), polydiacetylene (PDA), organic crystalline conductors based on molecular donors and acceptors of electrons. On the other hand, 1d-conductors are nontrivial systems. Thus, 1d-metal is unstable to the transition into semiconducting state. As a result the 1d-metal with half-filled conduction band becomes the Mott semiconductor or Peierls semiconductor. The Peierls transition leads to dimerization of the uniform regular 1d lattice (bond alternation) and semiconducting energy gap is proportional to the dimerization amplitude. The Mott transition is a result of electron correlation and energy gap in the Mott semiconductor vanishes with decreasing electron – electron interaction strength. The semiconductor of the Mott and Peierls type possesses some interesting properties. For example, the Mott semiconductors are characterized by antiferromagnetic structures [190], and in the Peierls semiconductors the kink-type excitations are possible [191, 192].
The influence of the Mott and Peierls instabilities on the properties of real quasi- one-dimensional systems have already long story. The main problem in theoretical studies consists in complications related to correct account of electron correlation effects. In ealier papers contradiction of the Mott and Peierls transitions was usually stated. Then it was shown that this contradiction is a result of one-electron approach in the RHF theory. The conclusion that the Mott and Peierls transitions coexist one with another was first made in [193]. This result was obtained due to more correct
526
treatment of pair electron correlations using varying localized geminals (VLG) approach [194 – 196]. It was shown that electron – electron interaction can enhance the Peierls dimerization [193]. This somewhat surprising result initiates several theoretical studies [197 – 202] which conformed the conclusion that even account for a small electron – electron interaction leads to increase in dimerization. This conclusion has been received on the basis of perturbation theory for infinite chains using computations [200] and the Feynman diagram technique [201]. Numerical calculations of short polyene chains within the same geminals approach conformed this result slightly deformed by boundary conditions [193].
Thus, we can state now that the theory predicts coexistence of the Mott and Peierls instabilities in real systems. So, the experimental data on 1d-systems should not correspond to the simple picture of the Peierls or the Mott semiconductors. One must expore the more complicated theoretical model including the both phenomena. On this way only one can give correct description of real 1d materials. For example, we can now give the correct answer to the question what mechanism of the forbidden gap formation is more essential – the electron correlation or dimerization.
In this paragraph we shall study now the simultaneous effect of the Mott and Peierls instabilities on electronic spectra and lattice distortion in real 1d conductors such as organic donor – acceptor molecular crystals and conjugated polymers of PA type. These studies are based on the VLG approach [193 – 196].
9.1. The Method of Calculations and Qualitative Evaluations
Studying the electronic properties of organic 1d materials the following model of uniform chain with the adiabatic Hamiltonian is used:
H = |
N |
|
|
+ cˆm+1,σ cˆmσ ) +γ ∑cˆm↑cˆm↑cˆm↓cˆm↓ |
+γ1∑nmnm+1 |
+ |
Kσ |
∑(xm − xm+1) |
|
,(301) |
||||
∑ βm (cˆmσ cˆm+1,σ |
2 |
|||||||||||||
ˆ |
|
|
+ |
+ |
+ |
+ |
|
|
|
|
|
|
||
|
σ,m=1 |
|
|
m |
|
|
m |
|
|
2 |
m |
|
|
|
where |
nmσ |
cmσ cmσ , |
number |
of sites N |
→ ∞ |
, |
xm is |
the |
mth |
site displacement, |
||||
|
|
|
= ˆ+ ˆ |
|
|
|
|
|
|
|
|
|
|
|
resonance integrals |
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
βm = −[β +(xm+1 − xm )β′] = −β(1+ ∆m ), |
β,β′> 0, |
|
|
(302) |
|||||||
γ and |
γ ′ are the electron repulsion parameters, Kσ is the lattice elasticity constant. |
|||||||||||||
Treatment below will be restricted by the most interesting case of half-filled conduction band with the number of electrons Ne = N . The Peierls deformation in this case reduces to the chain dimerization
x |
− x |
= |
(−1)m x , |
β |
m |
= −β[1+(−1)m |
∆] . |
(303) |
m+1 |
m |
|
0 |
|
|
|
|
|
The experimental values of displacements x0 are small as compared to the lattice |
||||||||
constant a. For example, |
in |
PA x0 |
= 0.07A and a = |
1.395 |
A [190, 203], for |
|||
527

[K+-TCNQ] complexes x0 = 0.18A and |
a = 3.6 A [204]. For small values of |
x0 the |
|
linear dependence |
|
|
|
∆ = β′ x |
(304) |
||
|
β |
0 |
|
is valid. The increase of displacement |
x0 → a |
destroys the relation (304) as well as |
|
the harmonic adiabatic approach used in (301). Thus, the method used here is valid only for small values of ∆ 1.
In this region Hamiltonian (301) is the Frohlich-type Hamiltonian with linear relative to displacements xm electron – phonon interaction.
Thus, when ∆ 1 the adiabatic approach is good enough and the problem of 1d instabilities is reduced to studying the ground state energy dependence on the value of ∆ (304). In other words, we need the ∆-value optimizing the expression
εt (∆) =εel (∆) + |
1 ∆2 |
, |
(305) |
||
2 |
к |
||||
|
|
|
|||
where εel is the electronic contribution into the ground state energy per an electron pair, and
к = (β′)2 / (2Kσ β) . |
(306) |
is the constant of electron – phonon interaction.
In order to calculate the electronic contribution into the ground state energy mentioned above the VLG approach will be used. The ground state wave function has the form
|
|
M ˆ |
+ |
|
|
|
|
|
M |
|
|
ˆ + ˆ + |
|
|
ˆ + |
ˆ + |
|
|
, |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
Ψ0 |
= ∏Gm |
0 |
|
≡ ∏(u fm↑ fm↓ |
+v fm↑ fm↓) |
0 |
|
|||||||||||||||||
|
|
m=1 |
|
|
|
|
|
|
m=1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
where |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ˆ |
|
|
|
|
ˆ |
|
|
|
|||
ˆ |
2 |
∑ |
|
ˆ |
|
|
|
−ikRm |
|
|
|
2 |
∑ |
|
−ikRm |
|
||||||||
fmσ = |
|
|
A |
e |
|
|
, |
fmσ |
= |
|
|
A |
e |
|
|
, |
||||||||
|
N |
|
|
|
N |
|
|
|||||||||||||||||
|
|
|
|
kσ |
|
|
|
|
|
|
|
|
|
|
kσ |
|
|
|
|
|||||
|
|
|
|k|<KF |
|
|
|
|
|
|
|
|
|
|
|
|
|
|k|<KF |
|
|
|
|
|||
|
|
ˆ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Akσ = akσ cosθk |
+ak |
σisinθk , |
|
|
|
|
|
|||||||||||||||
|
|
ˆ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Akσ = ak |
σ cosθk |
+akσisinθk , |
|
|
|
|
|
|||||||||||||||
|
|
|
|
a |
|
|
= |
1 |
|
|
N c |
e−ikna , |
|
|
|
|
|
|||||||
|
|
|
|
|
kσ |
|
|
|
|
|
|
nσ |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
N ∑n=1 |
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
u = cosϕ, |
v = sinϕ, |
|
|
|
|
|
|
|||||||||||||
2θk = arctan(λtgka), |
k = 2πl / Na, |
(l = 0,±1,±2,...) |
||||||||||||||||||||||
(307)
(308)
(309)
(310)
(311)
(312)
ϕ and λ are the variational parameters, the Fermi operators fˆmσ and fˆmσ correspond to the orbitals fmσ and fmσ which are partially localized near points
528

|
|
|
|
|
|
|
|
|
|
Rm = (2m +δ)a . |
|
|
|
|
|
(313) |
|||||||||
The ground state energy in units of β per electron pair has the form |
|
||||||||||||||||||||||||
|
|
|
|
εel = 2t cos2ϕ −k0 sin 2ϕ −v1 |
( |
2 |
∑| Pl |2 )cos2 2ϕ , |
|
(314) |
||||||||||||||||
N |
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
l |
|
|
|
|
|
|
|||||
where the kinetic energy average |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
t = ∑[1+(−1) |
m |
|
∆] fm (n) fm (n |
+1) |
= 0 |
|
ˆ |
ˆ |
+ |
|
0 , |
(315) |
|||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
fmσT |
fmσ |
|
||||||||||||||||
|
|
|
|
m |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
ˆ |
∑ |
+ |
+ σ +cˆ |
+ |
|
σ cˆmσ ), |
|
|
|
|
(316) |
||||||||
|
|
|
|
T = |
(cˆmσ cˆ |
+ |
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
m |
1, |
|
m |
1, |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
m |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
the exchange integral |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
K = 0 |
|
fˆm↑ fˆm↓Vˆee fm+↑ fm+↓ |
|
0 |
=U ∑| f0(n) |4 −U1∑| f0(n) |2 | f0(n +1) |2, |
(317) |
|||||||||||||||||||
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
n |
|
|
|
|
|
|
|
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
U =γ / β, U1 =γ1 / β, |
|
|
|
|
|
|
|||||||||||||
average of non-diagonal density or bond order |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
Pl = Ψ0 |
|
cˆl+σ cˆl+1,σ +cˆl++1,σ cˆlσ |
|
Ψ0 |
|
|
= ∑ fm (l) fm (l +1) = |
1 |
|
+(−1)l |
λ |
lnλ cos2ϕ . (318) |
|||||||||||||
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
4π |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
m |
|
|
|
|
|
π |
|
|
|
|
|||||
Now we consider the Hubbard approach γ1 = 0 in (301). Then, variation of the energy (314) with respect to ϕ gives
εel = −εg +U / 2, |
(319) |
where
εg = 
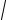 4t2 + K2 .
4t2 + K2 .
The values of t, K, P depend on the value of λ [193, 200], so
|
4 |
|
2 |
|
∂E(1−λ2 ) |
|
|
t(λ) = − |
|
E(1− x |
|
) +(4 −λ) |
|
|
, |
π |
|
∂λ |
|||||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
where the E(x) is the elliptic integral.
The explicit form of λ-dependence of K
K(λ) = U3 −Const λ lnλ.
(320)
(321)
(322)
can be obtained in the limit of small λ . We can see from (322) that when λ and, as a result, U are small the energy dependence (320) on λ is nonanalytic. Thus, we can suppose strong dependence of U on ∆0 which minimizes the total energy. Results of
529