

Учебники / Gene Therapy of Cochlear Deafness - Present Concepts and Future Aspects Ryan 2009
.pdf 78 Shou J, Zheng JL, Gao WQ: Robust generation of new hair cells in the mature mammalian inner ear by adenoviral expression of Hath1. Mol Cell Neurosci 2003;23:169–179.
78 Shou J, Zheng JL, Gao WQ: Robust generation of new hair cells in the mature mammalian inner ear by adenoviral expression of Hath1. Mol Cell Neurosci 2003;23:169–179.
 79 Staecker H, Praetorius M, Baker K, Brough DE: Vestibular hair cell regeneration and restoration of balance function induced by math1 gene transfer. Otol Neurootol 2007;28:223–231.
79 Staecker H, Praetorius M, Baker K, Brough DE: Vestibular hair cell regeneration and restoration of balance function induced by math1 gene transfer. Otol Neurootol 2007;28:223–231.
 80 Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Kelley PM, Kimberling WJ, Wagenaar M, LeviAcobas F, Larget-Piet D, Munnich A, Steel KP, Brown SDM, Petit C: Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995;374:60–61.
80 Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Kelley PM, Kimberling WJ, Wagenaar M, LeviAcobas F, Larget-Piet D, Munnich A, Steel KP, Brown SDM, Petit C: Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995;374:60–61.
 81 Weil D, Küssel P, Blanchard S, Lévy G, Levi-Acobas F, Drira M, Ayadi H, Petit C: The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 1997;16:191–193.
81 Weil D, Küssel P, Blanchard S, Lévy G, Levi-Acobas F, Drira M, Ayadi H, Petit C: The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 1997;16:191–193.
 82 Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ: Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 2001;68:26–37.
82 Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ: Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 2001;68:26–37.
 83 Wilson SM, Householder DB, Coppola V, Tessarollo L, Fritzsch B, Lee EC, Goss D, Carlson GA, Copeland NG, Jenkins NA: Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics 2001;74:228–233.
83 Wilson SM, Householder DB, Coppola V, Tessarollo L, Fritzsch B, Lee EC, Goss D, Carlson GA, Copeland NG, Jenkins NA: Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics 2001;74:228–233.
 84 Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K: Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 2001;27:103–107.
84 Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K: Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 2001;27:103–107.
 85 Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP: The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet 2001;27:99–102.
85 Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP: The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet 2001;27:99–102.
 86 Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER: Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 2001;69:25–34.
86 Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER: Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 2001;69:25–34.
84
 87 Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER: PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 2003;12:3215–3223.
87 Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER: PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 2003;12:3215–3223.
 88 Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C: A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 2000;26:51–55.
88 Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C: A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 2000;26:51–55.
 89 Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Lainé S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C: Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003;12:463–471.
89 Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Lainé S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C: Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003;12:463–471.
 90 Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Fauré S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P: A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 1997;15:186–189.
90 Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Fauré S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P: A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 1997;15:186–189.
 91 Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hördt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H: KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 1997;17:267–268.
91 Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hördt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H: KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 1997;17:267–268.
 92 Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ: KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 2000;97:4333– 4338.
92 Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ: KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 2000;97:4333– 4338.
 93 Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ: Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 2006;25:642–652.
93 Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ: Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 2006;25:642–652.
 94 del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, Menéndez I, Moreno F: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243–249.
94 del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, Menéndez I, Moreno F: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243–249.
 95 Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Söhl G, Willecke K, Chen P, Lin X: Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human con- nexin30-linked deafness. Proc Natl Acad Sci USA 2007;104:1337–1341.
95 Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Söhl G, Willecke K, Chen P, Lin X: Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human con- nexin30-linked deafness. Proc Natl Acad Sci USA 2007;104:1337–1341.
Kesser · Lalwani

 96 Kim JH, Auerbach JM, Rodriguez-Gomez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sánchez-Pernaute R, Bankiewicz K, McKay R: Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 2002;418:50–56.
96 Kim JH, Auerbach JM, Rodriguez-Gomez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sánchez-Pernaute R, Bankiewicz K, McKay R: Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 2002;418:50–56.
 97 Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R: Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001;292:1389–1394.
97 Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R: Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001;292:1389–1394.
 98 Li H, Roblin G, Liu H, Heller S: Generation of hair cells by stepwise differentiation of embryonic stem cells. Proc Natl Acad Sci USA 2003;100:13495– 13500.
98 Li H, Roblin G, Liu H, Heller S: Generation of hair cells by stepwise differentiation of embryonic stem cells. Proc Natl Acad Sci USA 2003;100:13495– 13500.
 99 Rivolta MN, Li H, Heller S: Generation of inner ear cell types from embryonic stem cells. Methods Mol Biol 2006;330:71–92.
99 Rivolta MN, Li H, Heller S: Generation of inner ear cell types from embryonic stem cells. Methods Mol Biol 2006;330:71–92.
 100 Wang Z, Jiang H, Yan Y, Wang Y, Shen Y, Li W, Li H: Characterization of proliferating cells from newborn mouse cochleae. Neuroreport 2006;17:767– 771.
100 Wang Z, Jiang H, Yan Y, Wang Y, Shen Y, Li W, Li H: Characterization of proliferating cells from newborn mouse cochleae. Neuroreport 2006;17:767– 771.
 101 Oshima K, Grimm CM, Corrales CE, Senn P, Martinez Monedero R, Géléoc GS, Edge A, Holt JR, Heller S: Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J Assoc Res Otolaryngol 2007;8:18–31.
101 Oshima K, Grimm CM, Corrales CE, Senn P, Martinez Monedero R, Géléoc GS, Edge A, Holt JR, Heller S: Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J Assoc Res Otolaryngol 2007;8:18–31.
 102 Li H, Liu H, Heller S: Pluripotent stem cells from the adult mouse inner ear. Nat Med 2003;9:1293– 1299.
102 Li H, Liu H, Heller S: Pluripotent stem cells from the adult mouse inner ear. Nat Med 2003;9:1293– 1299.
 103 Tateya I, Nakagawa T, Iguchi F, Kim TS, Endo T, Yamada S, Kageyama R, Naito Y, Ito J: Fate of neural stem cells grafted into injured inner ears of mice. Neuroreport 2003;14:1677–1681.
103 Tateya I, Nakagawa T, Iguchi F, Kim TS, Endo T, Yamada S, Kageyama R, Naito Y, Ito J: Fate of neural stem cells grafted into injured inner ears of mice. Neuroreport 2003;14:1677–1681.
 104 Okano T, Nakagawa T, Endo T, Kim TS, Kita T, Tamura T, Matsumoto M, Ohno T, Sakamoto T, Iguchi F, Ito J: Engraftment of embryonic stem cellderived neurons into the cochlear modiolus. Neuroreport 2005;16:1919–1922.
104 Okano T, Nakagawa T, Endo T, Kim TS, Kita T, Tamura T, Matsumoto M, Ohno T, Sakamoto T, Iguchi F, Ito J: Engraftment of embryonic stem cellderived neurons into the cochlear modiolus. Neuroreport 2005;16:1919–1922.
 105 Hu Z, Wei D, Johansson CB, Holmström N, Duan M, Frisén J, Ulfendahl M: Survival and neural differentiation of adult neural stem cells transplanted into the mature inner ear. Exp Cell Res 2005;302:40– 47.
105 Hu Z, Wei D, Johansson CB, Holmström N, Duan M, Frisén J, Ulfendahl M: Survival and neural differentiation of adult neural stem cells transplanted into the mature inner ear. Exp Cell Res 2005;302:40– 47.
 106 Corrales CE, Pan L, Li H, Liberman MC, Heller S, Edge AS: Engraftment and differentiation of embryonic stem cell-derived neural progenitor cells in the cochlear nerve trunk: growth of processes into the organ of Corti. J Neurobiol 2006;66:1489–1500.
106 Corrales CE, Pan L, Li H, Liberman MC, Heller S, Edge AS: Engraftment and differentiation of embryonic stem cell-derived neural progenitor cells in the cochlear nerve trunk: growth of processes into the organ of Corti. J Neurobiol 2006;66:1489–1500.
 107 Tamura T, Nakagawa T, Iguchi F, Tateya I, Endo T, Kim TS, Dong Y, Kita T, Kojima K, Naito Y, Omori K, Ito J: Transplantation of neural stem cells into the modiolus of mouse cochleae injured by cisplatin. Acta OtoLaryngol Suppl 2004;55:65–68.
107 Tamura T, Nakagawa T, Iguchi F, Tateya I, Endo T, Kim TS, Dong Y, Kita T, Kojima K, Naito Y, Omori K, Ito J: Transplantation of neural stem cells into the modiolus of mouse cochleae injured by cisplatin. Acta OtoLaryngol Suppl 2004;55:65–68.
108 Iguchi F, Nakagawa T, Tateya I, Kim TS, Endo T, Taniguchi Z, Naito Y, Ito J: Trophic support of mouse inner ear by neural stem cell transplantation. Neuroreport 2003;14:77–80.
109Hakuba N, Hata R, Morizane I, Yoshida T, Shudou M, Sakanaka M, Gyo K: Neural stem cells suppress
the hearing threshold shift caused by cochlear ischemia. Neuroreport 2005;6:1545–1549.
 110 Sharif S, Nakagawa T, Ohno T, Matsumoto M, Kita T, Riazuddin S, Ito J: The potential use of bone marrow stromal cells for cochlear cell therapy. Neuroreport 2007;18:351–354.
110 Sharif S, Nakagawa T, Ohno T, Matsumoto M, Kita T, Riazuddin S, Ito J: The potential use of bone marrow stromal cells for cochlear cell therapy. Neuroreport 2007;18:351–354.
 111 Jeon SJ, Oshima K, Heller S, Edge AS: Bone marrow mesenchymal stem cells are progenitors in vitro for inner ear hair cells. Mol Cell Neurosci 2007;34:59– 68.
111 Jeon SJ, Oshima K, Heller S, Edge AS: Bone marrow mesenchymal stem cells are progenitors in vitro for inner ear hair cells. Mol Cell Neurosci 2007;34:59– 68.
 112 Coleman B, Hardman J, Coco A, Epp S, de Silva M, Crook J, Shepherd R: Fate of embryonic stem cells transplanted into the deafened mammalian cochlea. Cell Transplant 2006;15:369–380.
112 Coleman B, Hardman J, Coco A, Epp S, de Silva M, Crook J, Shepherd R: Fate of embryonic stem cells transplanted into the deafened mammalian cochlea. Cell Transplant 2006;15:369–380.
 113 Iguchi F, Nakagawa T, Tateya I, Kim TS, Dong Y, Kita T, Kojima K, Naito Y, Omori K, Ito J: Surgical techniques for cell transplantation into the mouse cochlea. Acta OtoLaryngol Suppl 2004;551:43–47.
113 Iguchi F, Nakagawa T, Tateya I, Kim TS, Dong Y, Kita T, Kojima K, Naito Y, Omori K, Ito J: Surgical techniques for cell transplantation into the mouse cochlea. Acta OtoLaryngol Suppl 2004;551:43–47.
 114 Sakamoto T, Nakagawa T, Endo T, Kim TS, Iguchi F, Naito Y, Sasai Y, Ito J: Fates of mouse embryonic stem cells transplanted into the inner ears of adult mice and embryonic chickens. Acta OtoLaryngol Suppl 2004;551:48–52.
114 Sakamoto T, Nakagawa T, Endo T, Kim TS, Iguchi F, Naito Y, Sasai Y, Ito J: Fates of mouse embryonic stem cells transplanted into the inner ears of adult mice and embryonic chickens. Acta OtoLaryngol Suppl 2004;551:48–52.
 115 Sekiya T, Kojima K, Matsumoto M, Kim TS, Tamura T, Ito J: Cell transplantation to the auditory nerve and cochlear duct. Exp Neurol 2006;198:12–24.
115 Sekiya T, Kojima K, Matsumoto M, Kim TS, Tamura T, Ito J: Cell transplantation to the auditory nerve and cochlear duct. Exp Neurol 2006;198:12–24.
 116 Sugahara K, Shimogori H, Okuda T, Takemoto T, Yamashita H: Novel method for homogeneous gene transfer to the inner ear. Acta OtoLaryngol Suppl 2004;553:19–22.
116 Sugahara K, Shimogori H, Okuda T, Takemoto T, Yamashita H: Novel method for homogeneous gene transfer to the inner ear. Acta OtoLaryngol Suppl 2004;553:19–22.
 117 Kanzaki S, Beyer L, Karolyi IJ, Dolan DF, Fang Q, Probst FJ, Camper SA, Raphael Y: Transgene correction maintains normal cochlear structure and function in 6-month-old Myo15a mutant mice. Hear Res 2006;214:37–44.
117 Kanzaki S, Beyer L, Karolyi IJ, Dolan DF, Fang Q, Probst FJ, Camper SA, Raphael Y: Transgene correction maintains normal cochlear structure and function in 6-month-old Myo15a mutant mice. Hear Res 2006;214:37–44.
 118 Bedrosian JC, Gratton MA, Brigande JV, Tang W, Landau J, Bennett J: In vivo delivery of recombinant viruses to the fetal murine cochlea: transduction characteristics and long-term effects on auditory function. Mol Ther 2006;14:328–335.
118 Bedrosian JC, Gratton MA, Brigande JV, Tang W, Landau J, Bennett J: In vivo delivery of recombinant viruses to the fetal murine cochlea: transduction characteristics and long-term effects on auditory function. Mol Ther 2006;14:328–335.
 119 Liu Y, Okada T, Sheykholeslami K, Shimazaki K, Nomoto T, Muramatsu S, Kanazawa T, Takeuchi K, Ajalli R, Mizukami H, Kume A, Ichimura K, Ozawa K: Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol Ther 2005;12:725–733.
119 Liu Y, Okada T, Sheykholeslami K, Shimazaki K, Nomoto T, Muramatsu S, Kanazawa T, Takeuchi K, Ajalli R, Mizukami H, Kume A, Ichimura K, Ozawa K: Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol Ther 2005;12:725–733.
Inner Ear Gene Therapy and Stem Cell Transplantation |
85 |
 120 Boeda B, Weil D, Petit C: A specific promoter of the sensory cells of the inner ear defined by transgenesis. Hum Mol Genet 2001;10:1581–1589.
120 Boeda B, Weil D, Petit C: A specific promoter of the sensory cells of the inner ear defined by transgenesis. Hum Mol Genet 2001;10:1581–1589.
 121 Okano T, Nakagawa T, Kita T, Endo T, Ito J: Cellgene delivery of brain-derived neurotrophic factor to the mouse inner ear. Mol Ther 2006;14:866–871.
121 Okano T, Nakagawa T, Kita T, Endo T, Ito J: Cellgene delivery of brain-derived neurotrophic factor to the mouse inner ear. Mol Ther 2006;14:866–871.
 122 Maeda Y, Fukushima K, Nishizaki K, Smith RJ: In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet 2005;14:1641– 1650.
122 Maeda Y, Fukushima K, Nishizaki K, Smith RJ: In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet 2005;14:1641– 1650.
 123 Lalwani AK, Han JJ, Walsh BJ, Zolotukhin S, Muzyczka N, Mhatre AN: Green fluorescent protein as a reporter for gene transfer studies in the cochlea. Hear Res 1997;114:139–147.
123 Lalwani AK, Han JJ, Walsh BJ, Zolotukhin S, Muzyczka N, Mhatre AN: Green fluorescent protein as a reporter for gene transfer studies in the cochlea. Hear Res 1997;114:139–147.
 124 Staecker H, Brough DE, Praetorius M, Baker K: Drug delivery to the inner ear using gene therapy. Otolaryngol Clin North Am 2004;37:1091–1108.
124 Staecker H, Brough DE, Praetorius M, Baker K: Drug delivery to the inner ear using gene therapy. Otolaryngol Clin North Am 2004;37:1091–1108.
 125 Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP: Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 1998;125:557–566.
125 Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP: Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 1998;125:557–566.
 126 Melchionda S, Ahituv N, Bisceglia L, et al: MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 2001;69:635–640.
126 Melchionda S, Ahituv N, Bisceglia L, et al: MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 2001;69:635–640.
 127 Probst FJ, Fridell RA, Raphael Y, et al: Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene.[see comment]. Science 1998;280:1444–1447.
127 Probst FJ, Fridell RA, Raphael Y, et al: Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene.[see comment]. Science 1998;280:1444–1447.
 128 Wang A, Liang Y, Fridell RA, et al: Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3.[see comment]. Science 1998;280:1447–1451.
128 Wang A, Liang Y, Fridell RA, et al: Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3.[see comment]. Science 1998;280:1447–1451.
Anil K. Lalwani, MD
Department of Otolaryngology, New York University School of Medicine 550 First Avenue, NBV 5E5
New York, NY 10016 (USA)
Tel. +1 212 263 6344, Fax +1 212 263 8257, E-Mail anil.lalwani@nyumc.org
86 |
Kesser · Lalwani |

Ryan AF (ed): Gene Therapy of Cochlear Deafness.
Adv Otorhinolaryngol. Basel, Karger, 2009, vol 66, pp 87–98
Adenoviral and AAV-Mediated Gene Transfer to the Inner Ear: Role of Serotype, Promoter, and Viral Load on In Vivo and In Vitro Infection Efficiencies
Anne E. Luebkea Cherokee Rovab Peter G. Von Doerstenb David J. Poulsenb
aDepartments of Neurobiology and Anatomy and Biomedical Engineering, University of Rochester Medical Center, Rochester, N.Y., and bDepartment of Biomedical and Pharmaceutical Sciences, COBRE Center for Structural and Functional Neuroscience, University of Montana, Missoula, Mont., USA
Abstract
The lack of effective treatments for many forms of hearing and vestibular disorders has produced interest in virally mediated gene therapies. However, to develop a gene therapy strategy that would successfully treat inner ear disorders, appropriate viral vectors capable of transfecting cochlear and support cells must be identified. While virally mediated gene transfer into the inner ear has been accomplished using herpes simplex type I virus, vaccinia virus, retroviruses, adenovirus, and adenoassociated virus (AAV), we will restrict our discussion to AAV and adenoviral vectors. Issues such as vector toxicity and load, viral serotype and backbone, and promoter specificity are discussed and contrasted for both in vivo vs. in vitro inner ear gene transfer.
Copyright © 2009 S. Karger AG, Basel
Introduction
Virally mediated gene transfer into the inner ear has been accomplished using herpes simplex type I virus and vaccinia virus [1, 2], lentiviruses [3], retroviruses [4], adenoviruses [6–10] and adeno-associated virus (AAV) [11–13; see preliminary data]. Gene expression following transduction with lentivirus was restricted to cochlear cells lining the paralymphatic space [3]. In addition to the limited pattern of gene expression, lentivirus has the drawback of randomly integrating into the host chromosome. Adenoviral vectors have been used extensively to transfect cochlear and vestibular cells [5–10]. Adenovirus has been shown in vivo to transduce the scala tympani of the basal and second turn, over 90% of inner hair cells, more than 50%
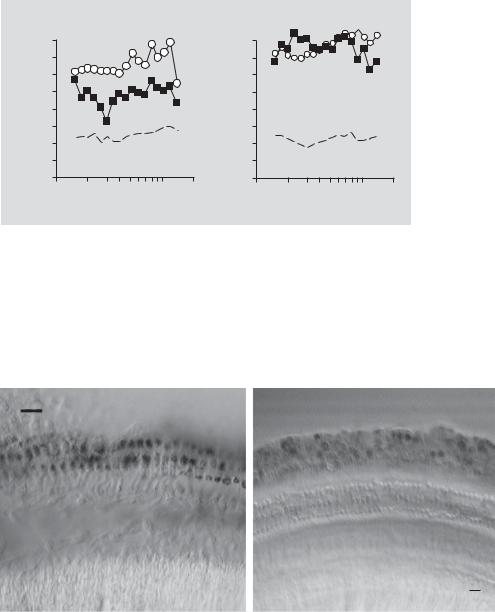
of outer hair cells, and some supporting pillar cells of the guinea pig [8, 10, 12]. In the mouse, adenovirus has been shown to transduce spiral ganglion cells and the epithelial lining of the scala tympani and Reissner’s membrane [9]. However, a significant drawback to using adenovirus as a gene therapy vector is that it introduces viral genes into the host cells that can stimulate an immune response. For instance, 1st-generation adenoviral vectors [E1–, E3–], have been shown to be toxic to cochlear hair cells in vivo [8] and in vitro [14]. In vivo, 1st-generation adenoviral vectors cause a severe decrease in distortion product otoacoustic emission (DPOAE) magnitudes as early as 48 h after viral infection, with outer hair cell death observed as early as 8 days after infection [12]. When 1st-generation adenoviral vectors were used to infect neonatal mouse cochlear or utricular cultures, hair cells survived and were transduced, yet their mechanosensitivity was compromised due to a loss of hair bundles [14]. However, when a 2nd-generation adenoviral vector had E1, E3, and E2b regions deleted, the neonatal and utricular hair cells infected with this modified adenoviral vector had intact hair cell bundles and robust mechanotransduction [15]. Moreover, when this 2nd-generation viral vector was infused into the guinea pig scala tympani (in vivo), hair cells were efficiently transduced and cochlear function (as assessed by DPOAE magnitudes) was not affected for 8–10 days after infusion [8]. However, by 28 days after infection, DPOAEs were reduced (fig. 1a). This reduction in DPOAE magnitudes could have possibly been due to a delayed immune response to the 2ndgeneration adenoviral vector. This conclusion is supported by the fact that when a gutted adenoviral vector was infused into the scala tympani of the guinea pig inner ear, DPOAEs were not reduced for up to 28 days (fig. 1b). This finding suggests that the loss of DPOAE amplitudes with 2nd-generation adenoviral vectors was due to a delayed immune response and this delayed immune response was eliminated by the use of gutted adenoviral vectors.
In addition to inner ear toxicity due to differences in the adenoviral vector backbone (1st vs. 2nd generation vs. gutted adenovirus), variations in adenoviral transfection efficiencies can occur due to slight differences in the promoter used to drive transgene expression. For instance, when the cytomegalovirus (CMV) promoter from human CMV was used to drive adenoviral-mediated transgene expression in the guinea pig inner ear, cochlear hair cells were predominantly transduced (fig. 2a). Whereas, when the CMV promoter from the murine CMV virus was used to drive transgene expression in the guinea pig inner ear, support cells (Hensen and Claudius cells) were predominantly transduced with no expression observed in cochlear hair cells (fig. 2b). This variability in transgene expression is similar to the mouse and human CMV replication patterns in the developing guinea pig fetus with human CMV replicating in epithelial cells and mouse CMV replicating in mesenchymal cells of the inner ear [16].
As discussed, effective in vivo infection depends of the use of the appropriate promoter to drive expression to the cochlear cell type of interest. Moreover, adenoviral titers used for in vivo infection (scala tympani infusion) must be 100× higher than
88 |
Luebke · Rova · Von Doersten · Poulsen |
|
[E1–,E3–, E2b–] adenovirus |
|
Gutted adenovirus |
|
SPL)- |
40 |
SPL)- |
40 |
|
30 |
30 |
|||
|
|
|||
(dB |
20 |
(dB |
20 |
|
10 |
10 |
|||
amplitude |
amplitude |
|||
0 |
0 |
|||
|
|
|||
DPOAE |
–10 |
DPOAE |
–10 |
|
–30 |
–30 |
|||
|
–20 |
|
–20 |
|
|
–40 |
|
–40 |
|
1.0 |
2.0 |
5.0 |
10.0 |
|
1.0 |
2.0 |
5.0 |
10.0 |
a |
Geometric mean frequency (kHz) |
b |
Geometric mean frequency (kHz) |
||||||
Fig. 1. Second-generation adenovirus infects the infused cochlea yet compromises cochlear function by 28 days after infusion. a Equi-level DPOAE measurements (L1 + L2 = 70 dB SPL; – – – = noise floor) are shown for perfused ears of 3 animals that received adenovirus at 5 × 109 pfu/cochlea. Compared with 30 min postoperatively ( ), by 28 days postoperatively ( ) there was a loss of DPOAEs in the infused cochlea. b In contrast, a gutted adenovirus infected the infused cochlea without compromising cochlear function by 28 days postoperatively. There was no significant change in the DPOAEs measured in the right (non-perfused ears; data not shown) for both viral vectors.
ohc3
ohc2
ohc1
ohc3
ohc2
ohc1
20 µm
a |
b |
Fig. 2. Photomicrographs of whole mounts of β-galactosidase-immunostained portions of guinea pig cochleae. a Adenovirus [E1–, E3–, and E2b–] infection with lacZ expression driven by the human CMV promoter stained outer hair cells (ohc) and inner hair cells (ihc) in the low-frequency regions of the cochlea. b Adenovirus infection with lacZ expression driven by the murine CMV promoter, however, stained Hensen and Claudius cells in the corresponding regions of the cochlea. All scale bars represent 20 μm.
Adenoviral and AAV-Mediated Gene Transfer |
89 |
those used for in vitro adenoviral infections. When neonatal mouse hair cells are transduced with adenoviral vectors, there are 2–3 orders of magnitude of viral titer load (dynamic range) over which hair cells are transduced, with half-maximal expression observed with a viral titer load of 107 pfu/endorgan [14]. For in vivo adenoviral infection, using the same promoter and viral preparation, viral loads of 109 pfu/endorgan [8] are needed to drive expression and the dynamic range between viral load and percent of hair cells transduced is very steep, spanning only one order of magnitude. In other words, whereas in vitro there is a broad dynamic range for adenoviral infection, in vivo this dynamic range is narrow and more restricted. However, a significant drawback to using adenovirus as a gene therapy vector is that adenoviral vectors introduce viral genes into the host cells that can stimulate an immune response. This results in the ultimate destruction of the transduced cells, toxicity, and a limited duration of transgene expression (as discussed earlier in review).
AAV has several characteristics which make it attractive as a gene delivery system [17–24]. AAV is a nonpathogenic human parvovirus that infects approximately 85% of humans within the first decade of life and has not been associated with disease. AAV has an extremely broad host range, capable of infecting most cell types, including post-mitotic cells. Recombinant AAV has demonstrated transduction and long-term gene expression in the liver, lung, muscle, brain, vasculature and retina of experimental animals [22, 25]. Furthermore, AAV vectors have been used in a number of clinical trials with no apparent pathological effects on cell growth, morphology or differentiation.
A number of advances in AAV production have improved the utility of recombinant AAV as a gene delivery vector. As a defective virus, AAV cannot replicate in the absence of helper virus. In the current packaging system for AAV, 96% of the viral genome is removed and helper virus is replaced with a plasmid-based system, completely eliminating the potential of contaminating helper virus [24, 26, 27]. The minimal 145-bp inverted terminal repeats are the only AAV viral DNA sequences retained (and the minimal cis elements necessary for packaging into AAV virions). Thus there are no viral genes present in the vector. This has the advantage of limiting the potential immunogenicity and toxicity of the vector. Recent improvements in the purification of AAV have enabled the large-scale production of highly pure virus stocks making the use of AAV more feasible for clinical trials. Original AAV purification methods relied on CsCl2 density gradient centrifugation [28]. Over time (24–72 h) CsCl2 reduces the infectivity of AAV. To circumvent this limitation, iodixinol gradient centrifugation combined with efficient column chromatography methods have been developed [29].
The identification of untranslated regulatory elements has lead to significant improvements in both the level and stability of transgene expression following AAV delivery. The woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) is a cis-acting enhancer that facilitates the expression of intronless viral messages [30, 31; see also preliminary data]. Our laboratory and others have
90 |
Luebke · Rova · Von Doersten · Poulsen |
demonstrated that insertion of the WPRE sequence between the stop codon and the poly-A sequence results in a substantial increase in both the stability and expression level of delivered transgenes [30, 31].
At least 10 different AAV serotypes have been identified based on amino acid sequence differences in their respective capsid proteins. Serotypes 1 and 6 share >99% amino acid homology and therefore are not functionally differentiated. AAV serotype 2 was the first to be cloned and therefore has been used in the vast majority of gene transfer studies to date and the only serotype examined in the auditory system. However, as the biological aspects and tissue tropisms of the other AAV serotypes have become better characterized, it is clear that the AAV serotypes 1–5 represent a rare resource for gene therapy applications. For example, serotypes 1, 2 and 3 require heparan sulfate proteoglycans as a co-receptor for viral entry into target cells. However, serotypes 4 and 5 do not use heparan sulfate proteoglycans as co-receptors. Indeed, it has been shown that AAV serotype 5 binds to sialic acid on the target cell surface [25]. This becomes relevant when attempting to target specific cell populations. For example, it was observed that AAV2 could not transduce human airway epithelial cells due to a lack of heparan sulfate proteoglycans. However, AAV5 efficiently transduced the same cells. Interestingly, significant differences in tropism and transduction efficiencies have been observed between serotypes 1 and 2, even though both depend on heparan sulfate proteoglycans as co-receptors. Transduction levels in the liver are 10to 50-fold higher for AAV2 compared to AAV1. In contrast, transduction of muscle tissue with AAV1 results in about a 100-fold increase in the production of the transgene product compared to AAV2 [32].
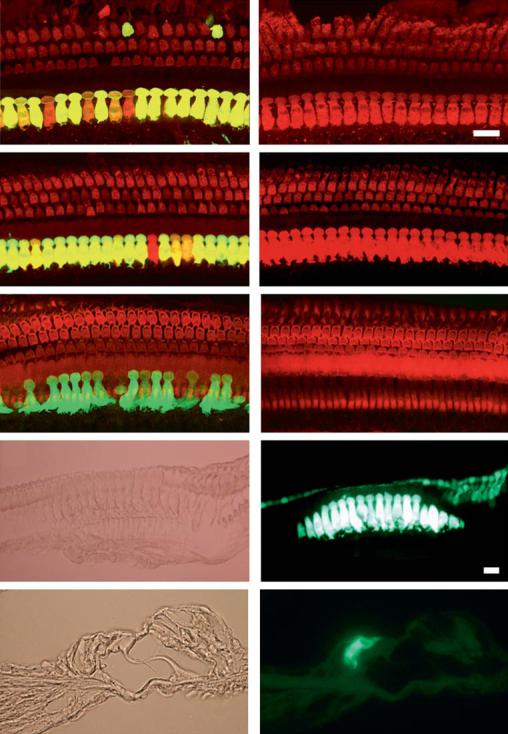
Our laboratory and others have reported serotype-specific differences in AAV transduction efficiencies within the cochlea, which may change depending on the model systems studied [33, 34, 35]. For example, we observed a high level of transduction within inner and outer hair cells in P0 cochlear explant cultures when AAV1 and AAV2 were used to deliver the eGFP transgene. However, under these same culture conditions, AAV5 failed to transduce cells within the organ of Corti [33]. In contrast, Liu et al. [35] observed transduction of only inner hair cells by AAV1, 2, 3, 5, 7 and 8 following direct injection into the mouse cochlea. While Bedrosian et al. [34] reported transduction of hair cells only with AAV1 and 8 following injection into P0 mouse pups and with AAV1, 2 and 8 following in utero injections. Using a mosaic AAV composed of both serotype 1 and 2 capsids, we have observed highly efficient transduction and expression of a GFP transgene primarily within the inner hair cells of adult guinea pigs (fig. 3) and adult mice (fig. 4) following direct injection into the scala tympani of the basal cochlear turn.
Transgene expression is dependent on viral serotype as well as the promoter used to drive transgene expression. Most of the early cochlear gene transfer studies relied on the human CMV immediate early promoter to drive transgene expression. The strong constitutive expression observed from this promoter in many cell types made it an obvious first choice. However, as we begin to improve gene delivery vectors
Adenoviral and AAV-Mediated Gene Transfer |
91 |
a |
b |
c |
d |
e |
f |
g |
h |
i |
j |
92 |
Luebke · Rova · Von Doersten · Poulsen |
it becomes necessary to examine alternative promoters that can provide cell typespecific expression. Fortunately there are a number of possible candidate promoters to choose from. Some of these promoters have been cloned and well-characterized, while others are currently being developed. Our laboratory and others have recently examined cochlear gene expression driven by the CAG promoter. CAG is a hybrid promoter, which contains sequence from the chicken β-actin promoter and the CMV promoter. Like the CMV promoter, the CAG promoter has demonstrated strong constitutive activity in multiple cell types. However, CAG promoter activity appears to be limited to hair cells within the mouse and guinea pig organ of Corti [33, 34–36] and is shown in figures 3 and 4. Boeda et al. [37] characterized the myosin VIIA promoter, which exhibits strong, selective expression in hair cells of the cochlea and vestibule. Recently, Lui et al. [36] further demonstrated that the myosin VIIA promoter provided selective expression of eGFP within hair cells following cochlear injection into adult rats and mice. These same authors also demonstrated that the neuron-specific enolase promoter and the elongation factor 1a promoter both provided selective eGFP expression within spiral ganglion cells and cells of the spiral ligament [36].
In some instances, support cells of the organ of Corti are targets for cochlear gene therapy manipulations [38]. Given that support cells exhibit an astrocytic-like phenotype, it is possible that astrocyte-specific promoters could provide support for cellspecific expression within the cochlea. More recently, the glial fibrillary acid protein (GFAP) promoter was shown to have selective activity within certain subpopulations of support cells. Rio et al. [39] observed GFAP activity in all supporting cells early after birth with an intensity gradient decreasing from the base towards the apex of the cochlea. We observed a similar pattern of GFP expression in AAV-transduced E13 explants grown for 5 days in culture when the GFAP promoter was used to drive eGFP expression (fig. 5).
We also examined the potential of the GFAP promoter to drive eGFP expression within support cells of the adult guinea pig cochlea injected with AAV1/2 vector (fig. 6). GFP expression was observed within what appeared to be Dieter’s cells as well as
Fig. 3. Guinea pig cochleae transduced in vivo with AAV using the CAG promoter. Merged whole mount images (60×) of a left ear (a) treated with 1011 genomic particles (gp) of AAV1-CAG-hrGFP virus and a right control ear (b). Whole mounts were stained with anti-myosin VIIa antibody (red). Under control of the CAG promoter, robust GFP expression was limited mostly to inner hair cells with only a few rare outer hair cells showing co-localization with myosin VIIa (yellow). Support cells in the organ of Corti do not show any evidence of GFP expression with the CAG promoter. Treated (c) and control ears (d) of an animal infused with 109 gp of the mosaic virus AAV1/2-CAG-hrGFP (3:1 ratio). GFP expression in inner hair cells is consistent although not uniform. Again GFP was found only occasionally in the outer hair cells (not shown) and is entirely absent in support cells. The stereocilia in e (treated the same as c) and f (control) are stained with phalloidin (red). g, h The same 40× images (g DIC; h GFP fluorescence) of 10-μm paraffin sections from the left ear of an animal treated with 1011 gp of the mosaic AAV1/2 virus. i (DIC), j (GFP fluorescence) 60× paraffin cross-sections from the same animal again showing primarily inner hair cell expression of GFP. All scale bars represent 20 μm unless otherwise noted.
Adenoviral and AAV-Mediated Gene Transfer |
93 |