

Неравновесные процессы в катализе
.pdfОбщая схема сопряжения для трехкомпонентной системы (см. выше):
A + In → X1 + jo , A + Acc → jo ,
1 → jo A + In → X 2 → jo .
Acc
Первая схема отражает брутто-превращения, а вторая – механизм действия промежуточного вещества Х.
Исходя из общей схемы, рассмотрим как пример реакцию сопряженного дегидрирования органического вещества пероксидом водорода.
Первичная реакция-распад пероксида водорода, при котором в систему генерируются активные центры:
Н2О2 + Н2О2 → [· HO2, · ОН] → 2H2O + O2.
актор индуктор интермедиаты
Вторичная реакция – дегидрирование:
Н2О2 + > CH – CH < → > C = C < + 2H2O
актор акцептор
Схематично это выглядит следующим образом:
|
|
|
|
|
|
2H2O O2 |
|
|
|
|
|
|
|
1 |
|
|
|
H2O2 H2O2 |
HO2 |
,OH |
2 |
C C 2H |
|
. |
||
|
|
|
|
|
|
2 |
O |
|
|
|
|
|
|
HC CH |
|
|
|
Недостаток такой схемы – |
излишняя формализация. Поэтому |
|||||||
более плодотворно раздельное описание механизма обеих реакций, при котором сохраняется индивидуальность каждой реакции и в то же время четко прослеживаются каналы связи посредством общих высокоактивных промежуточных веществ.
Первичная реакция:
< |
|
|
|
|
|
H2O2 → 2OH, |
|||||
< |
< |
|
|
|
|
H2O2 + OH |
→ H2O + HO2, |
||||
< |
< |
|
|
|
|
HO2 + “ Ë…*= → HO2 (=Ć“.) |
|
|
jo(H2O, O2 ). |
||
|
|
||||
< |
< |
|
|
|
|
|
|
|
|||
|
|
|
|
||
OH+ “ Ë…*= → OH(=Ć“.) |
|
|
|
|
|
|
|
|
|
||
|
|||||
H2O2 + H2O2 → 2H2O + O2. |
|||||
61

Вторичная реакция:
|
|
|
H2O2 |
OH H2O HO2, |
|
C C HO2 C C H2O OH.
Н2О2 +>CH–CH< → >C = C< + 2H2O.
Вторичная реакция по своей сути – окислительное дегидрирование. Это новый тип реакции – сопряженное окислительное дегидрирование, оно может быть осуществлено, в отличие от обычного термического дегидрирования, при более низких температурах, с более высокими выходами и селективностью. В результате химическойиндукцииизменяетсятипвторичной, целевойреакции. Ноесли она является самопроизвольной, то может не видоизменяться, а сохранять свой тип.
Возьмем нашу схему, но брутто-реакции разделим на элементарные стадии. Первичная реакция, согласно рассматриваемым представлениям, будет состоять из двух возможных процессов.
Первичная реакция:
A In X1 B In * , |
|
A In X1 B In * , |
X1 A In B1 ... |
или |
X 1 B1 ... |
|
|
|
2A In B In * B1 ... |
|
A In B In * B1 , |
где Х1 – промежуточная частица первого рода; In * – конечный продукт, образованный в основном из индуктора.
Вторичная реакция:
A In X1 B,
X1 Acc C ...
C Acc In B C ...
Здесь в качестве реагента присутствует индуктор. Отсюда видно, что индуктор, активируя актор, превращает его в такой промежуточный продукт первого рода, который представляет собой актив-
ный в химическом отношении фрагмент актора.
Актор, трансформируясьвХподвлияниемIn, переходитвновое состояние(активныйцентр) ивэтомкачествеучаствуетвпревращении акцептора.
62

Тогда начальная стадия первичной реакции
A InIn* X1.
Вторичная реакция:
A InIn* X1,
X1 Acc C ...
A Acc C ...
или же образуется высокореакционноспособный комплекс.
Первичная реакция:
A In X1In B, |
|
A In X1In B, |
X1In A B1 ... |
или |
X1In B1 ... |
|
|
|
2A In B B1 ... |
|
A In B B ... |
Вторичная реакция:
A In X1In B, X1In Acc C In.
A Acc C B.
5.2. Правила для определения механизма индуцированной реакции
Реакция, подвергающаяся индуцированию, изменяет свой тип, и в ее брутто-уравнение обязательно входят в качестве исходных реагентов вещества первичной реакции, из которых образовалась высокоактивная промежуточная частица (активный центр). Как правило, это актор. Вещество, способствующее генерированию активного центра (индуктора), но не входящее в его состав, не учитывается во вторичной реакции, в состав конечных продуктов вторичной реакции не входят продукты первичной реакции.
Сопряженные реакции имеют по крайней мере одну общую стадию.
Если использовать детерминанту, можно, не прибегая к методу квазистационарных концентраций Боденштейна, получить выражение, описывающее кинетику сопряженных реакций. За основу возьмем схему
63

k1 B ...
AIn,k0 X1
In*
Acc C ...
k2
Составим систему дифференциальных уравнений, учитывающих расход компонентов:
WIn d In 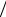 dt d A
dt d A 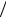 dt k0 A In , WC WAcc d Acc
dt k0 A In , WC WAcc d Acc 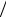 dt k2 Acc X ,
dt k2 Acc X ,
WB d B  dt k1 X .
dt k1 X .
Значения скоростей подставим в выражение для детерминанты
или |
D W |
|
W |
|
W |
|
W |
|
1 |
|||||
|
|
|
A1 |
|
Acc |
|
|
|
|
A2 |
|
Acc |
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
D |
1 k |
|
k |
2 |
|
Acc |
1 . |
|
||||
|
|
|
|
|
1 |
|
|
|
|
|
||||
Отношение k1 |
k2 |
Acc |
несет основную информацию об инду- |
|||||||||||
цирующем действии, |
количественно оценивает эффективность; k1 |
|||||||||||||
равно (или соизмеримо) с k2 |
Acc , |
D 1 2. Это равенство расхода |
||||||||||||
актора в обеих реакциях, расход акцептора также равен данной величине. Варьируя Acc , можно довести D до 1. Надо увеличивать концентрацию акцептора таким образом, чтобы k2 Acc k1 .
Еслипервичнаяреакциябимолекулярна, товчислителеотношенияпоявитсяпеременная, зависящаяотконцентрациилибоиндуктора, либо актора. Тогда увеличивается число возможных вариантов и, следовательно, явление химической индукции можно регулировать.
При химическом описании сопряженных реакций надо записать дифференциальные уравнения, учитывающие образование конечных продуктов в каждой из сопрягающихся реакций, и затем с помощью уравнения детерминанты получить соотношение, количественно оценивающее эффективность химической индукции. При этом не требуется использовать метод квазистационарных концентраций.
Перенос энергии в сопряженных реакциях происходит через общийпромежуточныйпродукт. Окисленнаяформапереносчикаболее богата энергией, чем восстановленная. Промежуточные вещества первого рода (высокореакционноспособные интермедиаты) и второго рода (медиаторы) являются исходными реагентами для последующих стадий.
64

5.3. Примеры применения сопряженных реакций. Проблема фиксации азота
Две формы минерального связанного азота – аммиачная и нитратная – непосредственно доступны растениям. Поэтому проблема фиксации азота в виде неорганических соединений весьма актуальна. Организм человека или животного лишен способности использовать для синтеза белка азот, входящий в состав неорганических соединений. Поэтому удовлетворение потребности человека и животных в белковых веществах зависит от того, в какой степени удовлетворяется потребность растений в минеральном связанном азоте. Разработанные методы связывания азота – дуговой, плазменный и т. д. не получили распространения, кроме синтеза аммиака.
Конечно, оптимальным было бы непосредственное окисление азота кислородом воздуха. Но как провести реакцию с минимальнымиэнергетическимизатратами? Можноосуществитьокислительную фиксацию азота. В системе N2 – H2O2 – H2O главным фактором, способствующимсвязываниюазота, являетсяхимическаяиндукция, проявляющаяся благодаря сопряжению реакций разложения перок-
сида водорода, который генерирует в систему вещество-посредник –
радикал HO2, передающий индуцирующее действие первичной реакции на реакцию окисления N2.
Механизм:
|
|
N2 HO |
2 N2O HO, |
N2 H2O2 N2O H2O.
N2 H2O2 N 2O H2O.
G2980 18 кДж/моль.
Слабое место предложенной схемы – относительно высокая эндотермичность, что ставит под сомнение возможность реализации этого реакционного пути на практике. Однако в результате химической индукции продукты индуцированной реакции могут образовываться с концентрациями, заметно превышающими термодинамически равновесные, что позволяет осуществлять химические процессы, равновесие которых сильно смещено в сторону исходных веществ.
65
Механизм:
N2O H2O H2N2O2, |
G = 170 кДж/моль |
||
N2O N2 O, |
|
(в условиях эксперимента). |
|
О – активный центр. |
|
||
|
|
|
|
O H2O OH |
OH, |
|
|
O H2 H2O2, |
G2980 (кДж/моль) |
||
N2O H2O2 N2O2 |
H2O, |
–22,9, |
|
N2O O N2O2, |
|
–131,3, |
|
|
|
–335,0. |
|
N2O HO2 N2O2 |
HO, |
||
N2O HO OH N2O2 OH HO N2O2 OH H2N2O2 O2,
G2980 141,4 кДж/моль.
N2O2 H2O2 H2N2O2 O2,
G2980 70,9 кДж/моль.
Здесь необходимо учитывать и вторичные реакции:
N2O2 O2 2NO2,
2NO2 H 2O HNO3 HNO2 .
Все вторичные реакции протекают при комнатной температуре, следовательно необходима закалка. Из приведенной схемы, анализируя полученные значения G стадий, можно сделать вывод о преодоленииэндотермичностиприпротеканииреакциипоэтомумаршруту и возможности ее применения для фиксации азота.
6. НЕКОТОРЫЕ АСПЕКТЫ НЕРАВНОВЕСНОГО КАТАЛИЗА
Причины неравновесности. Протекание химической реакции всегда приводит к нарушению равновесной функции распределения энергии и здесь имеются две главные причины. Первая связана с уменьшением концентрации богатых энергией, частиц реагентов вследствие того, что скорость их расходования за счет химической реакции существенно превышает скорость их пополнения. Вторая, приводящая к нарушению равновесного распределения энергии, связана с выделением энергии в ходе экзотермической реакции.
66

Эта энергия используется на возбуждение внутренних степеней свободы продуктов, а также на возбуждение твердого тела. В гетерогенном катализе в основном преобладает неравновесность второго типа.
Дезактивация возбужденных молекул – обязательная стадия многих процессовнаповерхности. Основной величиной, характеризующей обмен энергией между газовой фазой и поверхностью твердого тела, является средний тепловой коэффициент аккомодации (коэффициент Кнудсена)
|
|
E |
|
C=д |
E |
%2! |
(1) |
||
|
|
|
|
|
|
|
|||
E C=д E C%" |
|||||||||
|
|
|
|||||||
Для равновесного газа термический коэффициент аккомодации
|
|
(c R |
2 |
) c |
|
|
(2) |
||
|
t |
t |
|
|
r |
v |
, |
||
|
c |
c |
c |
R |
|
||||
|
|
|
|
|
|||||
|
|
t |
r |
|
v |
2 |
|
|
|
|
|
|
|
|
|
|
|||
где εt, εr, εv – парциальные коэффициенты аккомодации поступательной, вращательной и колебательной энергии соответственно; ct, cr, cv – парциальные мольные теплоемкости.
Вчислителеформулы(1) – разностьсреднихэнергийпадающих и отраженных частиц, а в знаменателе – разность средних энергий падающих частиц и этих частиц при полной термолизации с поверхностью.
Большинствоэкспериментовпроводитсявусловиях, когдапрактически вся энергия возбуждения сосредоточена либо на первом колебательном, либо на наиболее долгоживущем метастабильном электронно-возбужденном уровне и коэффициент аккомодации в этом случае приобретает особенно простой физический смысл – вероятностьпотериквантавозбужденияпри ударемолекулыо поверхность.
Вероятность образования возбужденных молекул при экзотермических стадиях каталитических процессов существенно зависит от механизма релаксации. Использование энергии, выделяющейся в экзотермических стадиях каталитического превращения, на ускорение этого процесса неоднократно обсуждалось в литературе, начиная с работ И. Лэнгмюра, который ввел понятие прекурсора, или предсорбционного состояния. Предположение о роли предсорбци-
67
онного состояния в катализе впервые было рассмотрено в работе С.З. Рогинского и Я.Б. Зельдовича в 1934 г. Для объяснения каталитическогоокисленияСОнаMnO2 онипредположилисуществование промежуточного, богатого энергией предсорбционного состояния молекулы СО, из которого она либо переходит в хемосорбированное состояние, неактивное для катализа, либо окисляется кислородом с
образованием СО2. |
|
|
|
|
СО(хем) |
CОгаз |
+ |
|
|
+ |
|
|
|
||||
|
|
|
|||
СОпредс |
|
СО2 |
|||
|
|
||||
|
|
|
|
|
|
При катализе, особенно селективном, энергия должна сосредоточиться на одной из связей реагирующей молекулы, чтобы обеспечить ее разрыв. Отсюда резонансные представления в катализе, которые появились в 1930-е гг. Тогда пытались доказать возможность подбора катализаторов по близости расположения отдельных линий в спектре испускания элементов, входящих в состав катализатора и реагирующих молекул и т. д.
О.В. Кобозев сформулировал принцип аггравации, согласно которому при данном строении активного центра каталитическая активность тем больше, чем больше молекулярная масса гомогенного катализатора. Эта закономерность объяснена способностью многоатомныхмолекулилитвердыхтелдлительноевремясохранятьэнергию возбуждения.
Уже в ХХ веке появились указания на сходство ряда закономерностейкатализаицепныхреакций. Так, внекоторыхкаталитических реакциях можно различить стадии инициирования, роста и обрыва цепи. Одним из первых на это обратил внимание С.З. Рогинский. В последующих работах было предположено, что в катализе возможно образование активных частиц типа радикалов, осуществляющих эстафетную передачу цепи; что в катализе возможно образование сверхравновесных концентраций активных частиц за счет использования энергии экзотермических актов реакции. Наиболее последовательно эти взгляды развивали Н.Н. Семенов и В.В. Воеводский, которые предположили, что за счет теплоты адсорбции молекулы на поверхности могут диссоциировать на поверхностные свободные радикалы (с вероятным участием свободных электронов твердого
68
тела). Эти свободные радикалы в дальнейшем участвуют в цепной реакции на поверхности катализатора.
Другая сторона вопроса о неравновесных процессах в катализе – перестройка поверхности и объема самого катализатора. Очевидно, что перестройка поверхности твердого тела и «каталитическая коррозия» – ускоренная перестройка катализатора в процессе катализа – также целиком обязаны энергии, выделяющейся в различных экзотермических процессах.
Само по себе наличие поверхности (обрыв решетки) вызывает изменение атомных расстояний в твердом теле вплоть до полной перестройки. Движущей силой этой перестройки является изменение энергетических уровней электронов на поверхности и как следствие этого изменение фононного спектра. Такая перестройка, изменяющая расстояние между атомами поверхности и конфигурацию активных центров, происходит во время катализа и адсорбции и существенно влияет на каталитическую и адсорбционную активность. Методом электронной микроскопии было показано, что на поверхности Pt и Pd под влиянием каталитических реакций происходит глубокое разрыхление. В работах А.Я. Розовского показано, что стационарный состав поверхности катализатора (работающего) может значительно отличаться от равновесного. При катализе происходит активация катализатора, которая связана со скоростью каталитической реакции: чем больше скорость каталитической реакции, тем больше производимые ею изменения поверхности катализатора.
При больших отклонениях от равновесия возникают так называемые диссипативные структуры. К ним принято относить упорядочение не только в пространстве, но и во времени. В гетерогенном катализе о наличии диссипативных структур свидетельствуют критические явления (резкое изменение скорости реакции при определенномзаполненииповерхности, определеннойтемпературеит. д.), множественность стационарных режимов, автоколебания и автоволны.
Использование понятия сродства реакции позволяет вывести условия необратимости протекания химической реакции. Сверхравновесные концентрации промежуточных веществ способствуют преодолению эндотермичности термодинамически невыгодных стадий. В условиях гетерогенного катализа возможен постоянный сдвиг
69
равновесия промежуточных стадий за счет их пространственного разделения, т. е. протекания разных стадий на пространственно разделенных активных центрах или даже на разных фазах. Кроме концентрационных градиентов, в гетерогенном катализе могут быть и температурные градиенты, сдвигающие промежуточные равновесия.
Вопрос о хемоэнергетическом стимулировании – это вопрос о нетепловом использовании сверхравновесной энергии химической реакции. Расчет показал, что при значительных энергиях возбуждения этот эффект может быть очень большим. Применительно к катализу расчет показывает, что образование возбужденных молекул на поверхности должно привести к увеличению выхода продуктов реакции. Энергетическое возбуждение способно передаваться поверхности, в результате чего в условиях катализа может образоваться сверхравновесная концентрация активных центров. Время релаксации этих центров намного больше времени жизни возбужденных молекул на поверхности. Поэтому основным каналом хемоэнергетического стимулирования в условиях гетерогенного катализа будет ускорение реакции в результате повышенной концентрации сверхравновесных активных центров при катализе. Расчет показывает, что в этом случае полная стационарность каталитической реакции достигается за очень большие времена, приближающиеся по порядку величины к времени работы катализатора.
Когда говорят, что сущность катализа заключается в протекании реакции по новому пути, через совокупность стадий с меньшей энергией активации по сравнению с некаталитической реакцией, то при этом не отражают все стороны явления. Не учитываются роль неравновесности активных центров в условиях катализа, образованиевкатализечастицсповышеннойсвободнойэнергией, сдвигпромежуточныхравновесийзасчетконцентрационныхииныхградиентов. Сродство реакции и ее стадии является более общим понятием, учитывающим не только энергетические, но и концентрационные изменения.
По И.Р. Пригожину, роль катализатора состоит в том, чтобы поддерживать очень высокое сродство между последним промежуточным и конечным продуктами. Эффективность системы увеличивается благодаря сильной неравновесности некоторых стадий. При
70