

Неравновесные процессы в катализе
.pdfне зависящая от их размера, скорость реакции обратно пропорциональна квадратному корню из среднего радиуса пор, а в области кнудсеновской диффузии, которая пропорциональна радиусу пор, скорость зависит от их среднего радиуса.
При уменьшении размера зерен катализатора, увеличении радиуса пор или снижении константы скорости (по сравнению с коэффициентом диффузии) концентрация вещества в глубине зерна становится не равной нулю и реакция переходит в область, промежуточную между внутридиффузионной и внутрикинетической. Для того чтобы за счет увеличения размера пор не слишком снижать внутреннюю поверхность зерен, благоприятна комбинация широких транспортных пор с микропорами. В промежуточной области работают многие промышленные катализаторы.
3. КИНЕТИКА КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Стационарный и квазистационарный режимы катализа
Вобщем случае стационарным является такой режим, когда
взаданном интервале времени в каждой точке реакционного пространства свойства системы не изменяются. Такими свойствами могут быть состав реакционной смеси, скорость реакции, состояние поверхности катализатора. Частным и предельным случаем стационарности является химическое равновесие в заданных условиях.
Понятие «стационарность» относится и к образованию, и к распаду промежуточных соединений. Для концентрации z каждого из промежуточных соединений в стационарном режиме соблюдается равенство
dzi / dt = 0, |
(1) |
означающее, что алгебраическая сумма скоростей образования и распадапромежуточныхсоединенийравнанулю. Еслисоставитьтакие равенства для каждого из промежуточных соединений, для линейных механизмов можно выразить концентрации промежуточных соединений через текущие концентрации реагентов, определяемые экспериментально, далее получить аналитическое выражение для скорости реакции и в первом приближении подтвердить или опро-
41
вергнуть предполагаемый механизм реакции. Это метод квазиста-
ционарных концентраций, или принцип Боденштейна (1913 г.).
Рассмотрим, например, реакцию А → В, протекающую на центрах Z через две обратимые стадии:
1.A + Z → AZ;
2.AZ → В + Z.
Обозначим концентрации реагентов через а и b, концентрации промежуточных веществ и свободных центров катализатора Z через z, а занятых центров AZ – через z1, константы скорости прямой и обратной реакций – через k1 и k–1, k2 и k–2 для первой и второй реакций соответственно. Общая концентрация всех центров z0 в стационарном режиме постоянна:
z + z1 = z0.
Тогда, используя принцип Боденштейна (1), напишем систему уравнений:
dz / dt k1az k 1z1 k2z1 |
k 2bz 0, |
(2) |
|
dz1 / dt k1az k 2bz k 1z1 k2z1 0. |
|||
|
|||
При условии равенства (1) решение дифференциальных уравнений заменяется на решение алгебраических уравнений. Из системы
(2) получаем
z |
k2 k 1 |
|
z0 , z1 |
|
|
k1a k 2b |
z0 . |
|
k1a k 2b k2 |
k 1 |
k2 |
k 2b k2 k 1 |
|||||
|
|
|
|
Промежуточные соединения, удовлетворяющие условию (1), иногданазываютбоденштейновскимипродуктами. Квазистационарный режим, при котором соблюдается условие (1), осуществляется, есливремяжизникаждогоизпромежуточныхсоединений(τ*) значительно меньше времени протекания реакции, т. е. необходимо, чтобы промежуточные соединения могли многократно образовываться
иразлагаться с сохранением постоянной концентрации во времени.
Впринципе, стационарность характерна для открытых систем (проточные реакторы). Однако принцип Боденштейна (1) применяли и для закрытых систем. Квазистационарный режим в них может осуществляться, если изменение концентрации промежуточных соединений в каждый момент времени будет отвечать условию (1) по отношению к так же изменяющимся концентрациям реагентов.
42
В условиях реакции может возникнуть такая ситуация, когда лишь часть промежуточных соединений удовлетворяет условию стационарности, а другая часть – это более долгоживущие соединения. Естественно, при этом снижается число дифференциальных уравнений типа (2). В системе останутся только уравнения для истинно боденштейновских продуктов.
Принцип квазистационарных концентраций Боденштейна широко использовался для получения кинетических уравнений и проверки гипотез о механизме реакции, о возможном участии в реакции того или иного промежуточного соединения. Основные сложности в примененииегодлякатализасвязанысотклонениямиконцентрации промежуточных веществ от постоянного их значения вследствие изменения активности катализатора, отравления поверхности, изменения внешних условий и т. д.
В последнее время принцип Боденштейна применяется для изучения механизма редко. Это вызвано тем, что в катализе (как и в гомогенной кинетике) появились физические методы, позволяющие прямо измерить промежуточные соединения вместо того, чтобы строить различные гипотезы об их образовании на основе принципа стационарности.
Закон действующих поверхностей. Формальным аналогом закона действующих масс в кинетике гетерогенно-каталитических реакций или, в общем случае, в кинетике процессов на твердых поверхностяхявляетсязакондействующихповерхностей. Этоттермин был предложен И. Лэнгмюром в 1918 г. применительно к реакциям частиц (молекул или атомов) на поверхности твердого тела, состоящей из определенного числа статистически расположенных адсорбционных центров, каждый из которых удерживает в адсорбированном состоянии одну частицу (молекулу или атом). Каталитическая реакция протекает за счет превращения этой молекулы на данном центре или за счет взаимодействия с другой молекулой на соседнем центре. При этом частицы, адсорбированные на соседних центрах, не взаимодействуют друг с другом (если они не вступили в реакцию). В общем случае в реакции участвуют как адсорбированные молекулы А, В и т. д. , так и свободные центры Z
1A + 2B + 3Z + ... → продукты
43
и для скорости каталитической реакции rK на адсорбционных центрах Z поверхности по закону действующих поверхностей можно написать
r |
k |
z 1 |
2 |
3 |
, |
(3) |
K |
s |
A |
B |
Z |
|
|
где z – общее число адсорбционных центров на единице поверхности; А, В – доля центров, заполненных адсорбированными веществами А и В; Z – доля свободных центров. Реакция протекает в мономолекулярном слое, поэтому А + В + Z + ... = 1. В частном случае закон действующих поверхностей описывает и процессы адсорбции, если 1 = 2 = 0, и десорбции, если 2 = 3 = 0.
Ограничений на применение закона действующих поверхностей к кинетике каталитических реакций еще больше, чем на применение закона действующих масс к кинетике гомогенных реакций. Во-первых, адсорбционный слой не является идеальным и предположение о равноценности всех центров не подтверждается экспериментами. Во-вторых, как правило, заполнения i экспериментально измерить трудно. Лишь в последнее время с появлением спектральных методов исследования in situ (которые могут применяться в условиях протекания каталитической реакции) стало возможным определение числа адсорбированных молекул. В-третьих, в реальных реакциях на поверхности, кроме заполнений i и концентрации свободных центров , в уравнение (3) часто входят также парциальные давления компонентов рi , когда молекула реагирует прямо из газовой фазы. Последние достижения в исследовании кинетики ге- терогенно-каталитических реакций показывают, что они могут протекать на границах островков, поверх прочносвязанного адсорбционного слоя, по типу цепных реакций с инициированием и обрывом.
4. КИНЕТИКА И МЕХАНИЗМ ЭЛЕМЕНТАРНЫХ АКТОВ НА ПОВЕРХНОСТИ
Теория абсолютных скоростей и ее применение к катализу
Теория активированного комплекса, теория (метод) переходного состояния, или теория абсолютных скоростей реакции, была создана в 1930-х годах Г. Эйрингом, М. Эвансом и М. Поляни. Она позволяет рассчитыватьскоростиэлементарныххимическихреакцийнаосновании знания об электронном строении и свойствах молекул реагентов.
44
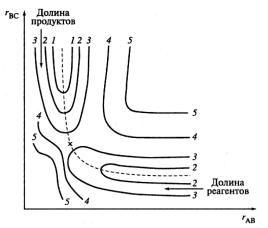
Поверхности потенциальной энергии
В основе теории абсолютных скоростей реакций лежит представление о многомерной поверхности потенциальной энергии реакционной системы. В общем случае многомерная поверхность потенциальной энергии системы из N атомов представляет функцию потенциальной энергии U от их внутренних координат или внутренних степеней свободы (вращательных и колебательных). В системе из N атомов число внутренних степеней свободы п = 3N – 6 (или п = 3N – 5, если ядра расположены на одной линии).
Простейшая двухмерная поверхность потенциальной энергии (п = 2) для реакции А + ВС → АВ + С при расположении атомов А, В и С на одной прямой изображена на рис. 4.
Рис. 4. Простейшая двухмерная поверхность потенциальной энергии для реакции А + ВС → AB + С при расположении всех трех атомов на одной прямой. По осям координат – межатомные расстояния rBC и rAB. Кривые 1…5 – уровни постоянной энергии; штриховая линия – координата реакции; × – седловинная точка
Реагентам и продуктам реакции на ней соответствуют относительно небольшие значения потенциальной энергии (долины), разделенные областью повышенной потенциальной энергии (потенциальный барьер). Штриховую кривую, проходящую по дну долин через потенциальный барьер, называют координатой реакции.
45
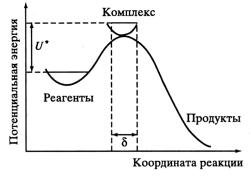
Часто используют одномерные схемы, представляющие сечение вдоль координаты реакции (рис. 5). На этих схемах состояния А + ВС и АВ + С являются устойчивыми минимумами, а вершине потенциального барьера соответствует седловинная точка, или точка перевала (×). Высота потенциального барьера определяется конфигурацией частиц, величиной энергии, необходимой для преодоления отталкивания, и некоторыми другими факторами. Каждому расстоянию между реагирующими частицами отвечает точка на поверхности потенциальной энергии.
Рис. 5. Профиль поверхности потенциальной энергии вдоль координаты реакции. Вертикальные линии ограничивают область активированного комплекса на координате реакции размером δ, горизонтальные – нулевые колебательные энергии реагентов и комплекса
Химическая реакция рассматривается как переход от конфигурации реагентов к конфигурации продуктов через точку ABC. Эту точку (или некий малый отрезок траектории реакции длиною ) на-
зывают активированным комплексом, или переходным состояни-
ем. Разность E0 между энергиями начального состояния и активированного комплекса ABC представляет собой энергию активации элементарной реакции А + ВС. Координата реакции – наиболее выгодный путь протекания реакции, требующий наименьших энергетических затрат.
Активированный комплекс
В основе теории активированного комплекса, или теории переходного состояния (она же – теория абсолютных скоростей) лежат три предположения.
46
1.Соблюдаетсямаксвелл-больцмановскоеравновесиемеждуак- тивированным комплексом и реагентами; поэтому их концентрацию можно вычислить с помощью функции распределения Максвелла – Больцмана.
2.Скорость реакции отождествляется со скоростью распада активированного комплекса. Реакция протекает с преодолением самого низкого потенциального барьера в точке активированного комплекса или вблизи от него.
3.Преодоление потенциального барьера вблизи активированного комплекса описывается как поступательное движение системы вдоль координаты реакции. Движение системы (протекание реакции) вдоль координаты реакции возможно только в направлении образования продуктов реакции. Это значит, что активированный комплекс, если уж он образовался, не может превращаться обратно
висходные вещества.
Это свойство коренным образом отличает активированный комплекс, описывающий элементарный акт реакции, от свойств промежуточныхпродуктов, описывающихпутьхимическогопревращения и обнаруживаемых физическими методами исследования. Уже самого образования активированного комплекса достаточно для осуществления реакции.
Активированные комплексы – это те же частицы или комплексы частиц, отличающиеся только конфигурацией с повышенным запасомэнергииинеустойчивыевнаправлениикоординатыреакции. Их среднее время жизни
* = 2 h / kT,
где h и k – постоянные Планка и Больцмана соответственно. При обычных для химических реакций температурах τ* = 10–13 с, т. е. близко к времени одного колебания. Такие времена стали доступны экспериментатору относительно недавно, с появлением фемтосекундной спектроскопии (фемто – 10–15), в которой для идентификации частиц применяют лазеры с импульсами длительностью до 10–14 с, т. е. меньше времени одного колебания.
Теория абсолютных скоростей для реакций на поверхности
Активированный комплекс принимается адсорбированным на поверхности с одной существенной особенностью. Она заключается
47
в том, что необходимо учитывать возможность занятия нескольких центров поверхности одним активированным комплексом. Кроме того, в отличие от гомогенных реакций активированные комплексы наповерхности, какправило, необладаютпоступательнымивращательным движением.
Концентрация активированных комплексов на поверхности должна зависеть от числа центров, занятых активированным комплексом, от числа способов осуществления конфигурации активированного комплекса, от его структуры, структуры поверхности, параметра решетки твердого тела, размеров молекулы, общего числа центров на поверхности.
Рассмотрим общий случай для элементарной реакции на поверхности
aA + bB + x(ZC) + y(ZD) + zZ → продукты.
Здесь А и В – вещества, реагирующие из газовой фазы; С и D – вещества, вступающие в реакцию из адсорбированного состояния; Z – свободные центры; а, b, х, у и z – стехиометрические коэффициенты; z – число свободных центров, необходимых для участия в реакции. В действительности в элементарной реакции участвует не более трех частиц, считая и свободные центры. Следовательно, a + b + x + y + z = 3 и некоторые из членов левой части равны нулю.
Согласно теории абсолютных скоростей реакций константа скорости реакции на поверхности
k |
(kT /h)N (gF /F a F b F x |
F y |
)e E0 /kT , |
(4) |
c |
A B ZC |
ZD |
|
|
где χ – трансмиссионный коэффициент, который учитывает возможность того, что не каждый активированный комплекс, достигший вершины барьера и движущийся по координате реакции, действительно будет распадаться и давать продукты реакции; N – число центров на единице поверхности; g – число возможных расположений активированного комплекса на поверхности (если фиксирован один конец активированного комплекса, g = 1; если активированный комплекс занимает два центра, g = 2, 4 или 6 в зависимости от симметрии решетки поверхности); F – статистическая сумма активированного комплекса без вклада координаты реакции; FA, FB, FZC и FZD
– статистические суммы веществ А, В, ZC и ZD, рассчитанные из энергий микросостояний, отсчитанных от нулевой энергии данной
48
частицы в газовой или адсорбированной фазе; E0 – энергетический барьер реакции (энергия активации при Т = 0).
Пользуясь уравнением (4), можно найти размерность и порядок величиныk0 простейшихгетерогенныхреакций, которыемогутбыть элементарными. Эти размерности, отнесенные к одному центру поверхности, приведены в табл. 1. Адсорбционный коэффициент выражается соответственно в см3/молек.
Сопоставляя экспериментальные значения k0 с расчетными из табл. 1, можно определить, по какому механизму протекает реакция.
Таблица 1
Размерность и интервалы изменения предэкспоненциальных множителей для простейших гетерогенных реакций
|
Гетерогенная реакция |
Размерность |
Порядок |
|
константы скорости |
величины ka |
|
|
|
||
1. |
Одноцентровая адсорбция атомов |
см3/(ат · с) |
10–11–10–12 |
2. |
Одноцентровая адсорбция молекул |
см3/(молек · с) |
10–11–10–15 |
3. |
Диссоциативная двухцентровая |
см5/(молек · с) |
10–28–10–30 |
адсорбция линейных молекул |
|
|
|
4. |
Диссоциативная двухцентровая |
см3/(молек2 · с) |
10–39–10–42 |
адсорбция нелинейных молекул |
|
|
|
5. |
Мономолекулярная десорбция |
с–1 |
1013 |
6. |
Рекомбинационная десорбция |
см2/(молек · с) |
10–1–10–8 |
7. |
Поверхностная миграция атомов |
с–1 |
1013 |
8. |
Поверхностная миграция молекул |
с–1 |
1010–1013 |
9. |
Мономолекулярная реакция |
с–1 |
1010–1013 |
в адсорбированном слое |
|
|
|
10. Бимолекулярная реакция по |
|
|
|
ударному механизму (механизм |
см3/(молек · с) |
10–10–10–19 |
|
Или – Ридила) |
|
|
|
11. Бимолекулярная реакция в |
|
|
|
адсорбированном слое (механизм |
см2/(молек · с) |
10–1–10–8 |
|
Лэнгмюра – Хиншельвуда) |
|
|
|
12. Рекомбинация радикалов с |
|
|
|
участием «стенки» как третьей |
см2/(молек · с) |
10–33–10–47 |
|
частицы |
|
|
|
49
Число активных центров
Число активных центров поверхности можно оценить, разделив экспериментальное значение k0 на рассчитанное по формуле (4) по теории абсолютных скоростей. Общий результат таких расчетов для разных каталитических реакций сводится к тому, что число активных центров получается значительно меньшим, чем число атомов поверхности (1015 см–2). При этом на металлах число активных центров сравнительно велико и приближается к числу поверхностных атомов металла. Возможно также, что экспериментальные значения k0 занижены не только из-за малого числа активных центров, но и вследствие низких значений трансмиссионного коэффициента .
Границы применения теории абсолютных скоростей
Теория абсолютных скоростей применима, только если считать изучаемые процессы адиабатическими (адиабатическое приближение). В данном случае подразумевается, что система, пришедшая в переходное состояние, неизбежно перейдет в конечное состояние только при плавном изменении всех параметров, т. е. при плавном перемещении точки, изображающей состояние системы, вдоль поверхности потенциальной энергии. Степень отклонения от адиабатичности принято учитывать трансмиссионным коэффициентом . Последний включает в себя также конечное значение вероятности туннельного перехода, т. е. перехода в область продуктов в случае системы, имеющейэнергиюниженулевойэнергииактивированного комплекса. Количественный учет неадиабатичности и туннельного эффекта в настоящее время возможен лишь для простейших моделей газовых реакций.
В теории абсолютных скоростей не рассматривается процесс образования активированных комплексов. Вместо этого принима-
ется, что их концентрация соответствует равновесному распределению Максвелла – Больцмана. Не рассматривается в этой теории и дальнейшая судьба реагирующей системы атомов после пересечения потенциального барьера. Считается, что такой переход автоматически приводит к образованию продуктов элементарной реакции. Оба эти допущения имеют свои границы применимости и за их пределамиизменяетсянетольковыражениедляконстантыскорости, но и общий вид кинетического уравнения.
50