

Неравновесные процессы в катализе
.pdfэтом скорость производства энтропии чрезвычайно возрастает. Поэтому можно представить себе и такие случаи, когда энергия активации многостадийной каталитической реакции (а в сущности, все каталитические реакции многостадийны) будет не ниже энергии активациинекаталитическойреакции. Однакореакциябудетпротекать с большей суммарной скоростью и селективностью за счет сдвига промежуточных равновесий.
Сейчас имеется не очень много достоверных данных о влиянии энергииисходныхреагентовнапротеканиекаталитическойреакции, однакопообратномупроцессу– распределениюэнергиивпродуктах каталитической реакции – таковых достаточно. Наиболее естественноизучатьэтораспределениевпродуктахэкзотермическойреакции.
Простейшей высокоэнергетической каталитической реакцией является рекомбинация атомов с образованием двухатомной молекулы
А + А → А2.
Именнонапримереэтойреакциибыливпервыеобнаруженынеравновесные эффекты уноса энергии. Например, исследование процессов рекомбинации атомов методом молекулярных пучков показало, что вылетающие с поверхности монокристаллов Ni, Cu, Pt молекулы H2 действительно более «нагреты», чем поверхность. Этим же объясняется и голубое свечение, наблюдаемое при пропускании атомов азота с добавкой атомов кислорода над поверхностями Co, Ni, Cu; при этом образуются возбужденные молекулы NО.
Высокоэнергетическими являются также реакции каталитического окисления, поэтому и здесь в последнее время широко изучают возможность уноса части энергии реакции ее продуктами. Многочисленные эксперименты, например по окислению СО на Pt, показали, что с поверхности десорбируются «горячие» молекулы СО2. Отмечено, что в газофазных реакциях неравновесные эффекты важны при Ea / RT < 5÷10.
При изучении разложения NО на поликристаллической Pt из термодесорбционных спектров следует, что продукт разложения N2 выделяется при несколько более высокой температуре, чем NО, а кислород – при значительно более высокой. В общем случае температура возбужденных (поступательно и колебательно) частиц, вылетевших с поверхности при катализе, столь велика (больше 1000 К),
71
что весьма вероятны их дальнейшие реакции в приповерхностном слоегазаукатализатора. Однакоработы, гдебыявлениекатализарассматривалось с этой точки зрения, неизвестны. Значительные отклонения от равновесного распределения энергии в молекулах – продуктахкаталитическойреакцииуказываютнанеравновесностьнетолько последней стадии процесса (десорбции из слабосвязанного состояния), но и предыдущих стадий. По принципу микрообратимости колебательное, поступательное и вращательное возбуждение исходных реагентов также должно оказывать существенное влияние на катализ.
Итак, существуют два механизма дезактивации возбужденных частиц при адсорбции и катализе: 1) неупругое рассеяние, прямая адсорбция; 2) взаимодействие типа захват – десорбция, непрямая адсорбция. В процессах второго рода постулируется существование на поверхностинекоегопромежуточного, предсорбционногосостояния, или прекурсора. Это состояние может мигрировать по поверхности.
Основными экспериментальными доказательствами считали данные о зависимости коэффициента прилипания S при адсорбции от степени заполнения Ө. Если бы адсорбция протекала по закону Лэнгмюра с последовательным статистическим заполнением адсорбционных центров, то наблюдалась бы линейная зависимость S от (1 – Ө). На самом деле часто наблюдается практическая независимость S от Ө. Очевидно, образуется прекурсор – предсорбционное состояние адсорбированной молекулы, когда она может свободно двигаться по всей поверхности, в том числе над хемосорбированными молекулами, вплоть до прочной хемосорбции или каталитической реакции. В дальнейшем такой прекурсор, который может существовать над хемосорбированными молекулами, назвали внешним (extrinsic) прекурсором. Если же прекурсор – предшественник хемосорбции существует лишь на свободных центрах адсорбента, он называется внутренним (intrinsic) прекурсором.
6.1. Энергетическая схема адсорбции с прекурсором
Из состояния с прекурсором молекула может перейти в прочно хемосорбированное состояние с энергией активации Еа (рис. 7). Существуютдваосновныхобъясненияприродыпрекурсора: а) прекурсор есть физически адсорбированная молекула; б) прекурсор есть неравновесное, колебательно-возбужденное состояние.
72
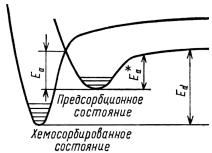
Рис. 7. Потенциальные кривые образования предсорбционного состояния при хемосорбции
Прямые доказательства существования прекурсора: реально наблюдали методом рентгенофотоэлектронной спектроскопии (РФЭС) физически адсорбированный СО на Cu при 20 K. При 30 К до 40 % СО десорбировалось, а 60 % превращалось в молекулярно-хемосор- бированное состояние, которое было стабильно свыше 100 К. Также наблюдали аналогичное превращение физически адсорбированного О2 наAl (111) воксидныйслойисуществованиефизическиадсорбированного прекурсора для хемосорбции N2 на Ni (100), Ru (001), W (100). Вэтихслучаяхфизическаяадсорбцияестьпрекурсор, энергия активации хемосорбции из которого ниже, чем Еа хемосорбции из газовой фазы.
Но есть и работы, указывающие на неравновесную природу прекурсора. Экспериментально показано, что захваченная поверхностью молекула может совершить десятки и сотни элементарных скачков до прочной хемосорбции. Кроме того, на разных гранях одного и того же монокристалла наблюдаются разные зависимости S(Ө), что маловероятно при физической адсорбции. Некоторые эксперименты заставляют предположить, что молекулы в предсорбционном состоянии могут быть колебательноили электронно-возбуж- денными. Так, N2 при адсорбции на W находится в возбужденном состоянии, а затем последовательно теряет свою энергию при скачках по поверхности с последующей хемосорбцией. Эксперименты с молекулярными пучками демонстрируют уменьшение S с ростом Еi – поступательной энергии молекул в пучке. Для прямой адсорб-
73
циихарактернасильнаязависимостьS отТi, дляадсорбцииспрекурсором – зависимость S от ТS. В работе П. Эрлиха (1958) предложена формальная модель для адсорбции молекулы А через состояние прекурсора Апр
A |
S * |
|
kaω(Θ) |
, |
→ ` |
→ A |
|||
ą=ƒ |
← |
C! |
.Ëä |
|
|
k* |
|
|
|
|
g |
|
|
|
где Ахем – хемосорбированное состояние; ka ( ) – константа скорости |
||||
перехода из прекурсора в хемосорбированное состояние, пропорциональная вероятности ω(Ө) найти вакантное место в хемосорбированном слое. В квазистационарном приближении:
S k ( )n* S |
|
|
(1 K ) |
, |
(3) |
||
0 [1 K / ( )] |
|||||||
a |
|
|
|
||||
K k* / k , S |
0 |
|
S * / (1 K ). |
(4) |
|||
g a |
|
|
|
|
|
||
Далее: показано (1978), что окисление NH3 протекает на реберных атомах ступенчатой поверхности Pt. Однако скорость окисления NH3 на этих атомах примерно на порядок больше числа ударов молекул о те же атомы. Был сделан вывод, что кислород находится в виде подвижного прекурсора на плоскости Pt и затем диффундирует к реберным атомам. В 1981 г. показали, что окисление Н2 на Pt протекает через возбужденный прекурсор Н*:
Н2 → Н*адс + Надс; Н*адс + Оадс → ОНадс; ОНадс + Н*адс → Н2О.
В1986 г. состоялась международная конференция по кинетике граничных реакций, на которой обсуждался вопрос: «Прекурсор: миф или реальность?» Сейчас ясно, что это реальность.
6.2.Хемоэнергетическое стимулирование
В1980 г. была предпринята попытка теоретически выяснить, возможно ли использование энергии, выделяющейся в одной реакции, для прямого ускорения другой. Среди необходимых условий указаны следующие: а) вторая реакция должна быть неравновесной (под неравновесностью в данном случае подразумевается обеднение возбужденного состояния в ходе реакции, когда вероятность превращения из возбужденных состояний намного больше, чем веро-
74
ятность релаксации); б) энергия, выделившаяся в первой реакции, должна превышать энергию активации второй реакции.
О.В. Крылов и Б.Р. Шуб совместно с А.Я. Розовским проанализировали некоторые простейшие кинетические модели нетеплового стимулирования химической реакции и определили условия, при которых эффективность стимулирования достаточно велика.
Хемоэнергетическое стимулирование (ХЭС) – это стимули-
рование химической реакции путем нетеплового использования сверхравновесной энергии продуктов другой реакции. Под стимулированием обычно понимается изменение каких-либо параметров химической реакции для достижения желаемого результата. Для нас стимулирование – получение возможно большего выхода продуктов реакции за один и тот же промежуток времени t с начала ее проведения, равно как и снижение параметров процесса (температура, время) при сохранении величины выхода целевого продукта. Исследование влияния стимулирующего воздействия должно проводиться при прочих равных условиях.
СогласнотрактовкенаданныймоментвременимеханизмыХЭС сводятся к двум случаям.
1.Энергетически неравновесные продукты одной реакции являются исходными или промежуточными веществами другой реакции. Так как при этом первая (стимулирующая) реакция представляет собой генератор реагентов второй (основной) реакции, естественно называть эту ситуацию генерационным стимулированием.
2.Неравновесные продукты стимулирующей реакции могут обмениваться энергией с исходными или промежуточными веществами основной реакции. В этом случае можно говорить об обменном стимулировании.
Вгетерогенном катализе возможно образование активных частиц: свободных атомов или радикалов, вылетающих с поверхности
вгазовую фазу, квазисвободных частиц, ведущих цепную реакцию
вдвухмерном адсорбционном слое, а также возбужденных молекул. Однако наиболее распространенным случаем является образование неравновесных концентраций активных центров самого катализатора, аналогичныхактивнымчастицамцепнойреакциивгазовойфазе.
Рассмотрим простейшую двухцентровую схему для реакции
А → В:
75
A Z k1 A' Z* ,
A Z* k2 B Z*,
* k3
Z Z,
k#3
где Z – активный центр, k1 – константа скорости активации катализатора реагентом А; k2 – константа скорости катализа; k3 – константа скорости дезактивации активного центра; k–3 – константа скорости термического возбуждения активного центра.
Первая стадия аналогична стадии инициирования в цепной реакции, вторая – аналогична стадии продолжения цепи, третья – стадии линейного обрыва цепи. Наиболее распространены в катализе механизмы, когда процесс протекает на центрах разной активности
инаблюдается активация в ходе самого процесса. Центры Z были раньше, центры Z* – возникают в ходе катализа. На центрах Z* реакция протекает быстро, на Z – медленно. Часто говорят, что отличие катализаотцепныхреакцийзаключаетсявтом, чтовкатализеактивные центры существуют до начала реакции, а в цепных реакциях – образуются после инициирования реакции. Однако в рассмотренных выше системах до реакции существуют малоактивные центры Z, а в процессе катализа образуются другие, более активные центры Z* (нерелаксированные, или возбужденные центры), на которых и протекает каталитическая реакция. Здесь различие между катализом
ицепной реакцией менее существенно.
Один из доказанных цепных механизмов катализа – окисление СО на нанесенном катализаторе V2O5/SiO2:
2V5+ + O2– + CO → CO2 + 2V4+,
V4+ + O2 → V5+ + O2–,
V5+ + O2– + V4+ → 2V5+O–,
2V5+O– + CO → CO2 + V4+,
V4+O2– + V4+→ O2– + 2V5+.
Здесь первая стадия аналогична инициированию цепи, она приводит к образованию активных центров; дальше – развитие цепи, когда роль активных промежуточных частиц играют как центры V4+, так и радикалы кислорода O– и O2– (или V5+O– и V5+O2–), а последняя
76
стадия – регенерация исходной поверхности – играет роль обрыва цепи. Стационарная концентрация адсорбированных радикалов кислородасущественновышеравновесной. ОкислениеСОосуществляется за счет сопряжения с реакцией восстановления поверхности катализатора.
Поверхностные цепи образуются и в многочисленных реакциях полимеризации. На примере синтеза углеводородов на железных и кобальтовых катализаторах А.Я. Розовским было изучено другое явление, сближающее катализ и цепные реакции, – явление кинетического сопряжения. Было показано, что стационарный состав железногокатализаторавусловияхкатализазначительноотличается от равновесного, причем эти отклонения определяются протеканием каталитической реакции. Как в цепных реакциях, так и во время катализа при определенных соотношениях скоростей стадий могут возникать неравновесные концентрации промежуточных частиц и создается возможность кинетического сопряжения, когда термодинамически затрудненные стадии становятся возможными.
Итак, признаки, характерные для цепной реакции и наблюдающиеся часто в катализе: а) чередование образования и гибели активных центров (заполнение центров на поверхности и их освобождение); б) образование сверхравновесных концентраций промежуточных веществ, способствующее преодолению эндотермичности невыгодных стадий; в) достижение максимальной скорости через некоторое время после начала реакции.
Если химическая реакция в системе является единственным процессом, изменение энтропии в ней связано с изменением числа молей компонентов:
dS # |
1 |
i dni |
# |
1 |
i id 0. |
(5) |
|
T |
T |
||||||
|
i |
|
i |
|
Здесь ξ = ni / νi – степень протекания реакции (число превратившихся эквивалентов).
Сродствореакции Ai # i i (введеноТ. деДонде). Ai = dG/dξ.
i
Суммарное сродство реакции Ai Sik Aik , где Sik – стехиометрическое число стадий k в реакции i. k
Среднее стехиометрическое число
77
|
|
|
|
|
Sik Aik |
|
|
Ai |
|
|
i i |
dG / d , |
(6) |
||||
|
|
S |
k |
|
|
, Ai |
|||||||||||
|
|
Aik |
Ai |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
i |
|
|
|
|
|
|||
|
|
|
|
|
i |
|
|
|
i |
|
ξ = ni / νI. |
|
|
|
(7) |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Обобщенное уравнение де Донде ri |
exp(A |
|
|
– из его |
|||||||||||||
SRT ) |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
r |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
i |
|
|
|
|
|
же уравнения ri r exp(Ai RT ) . Вблизи от равновесия A SRT 1 |
|||||||||||||||||
|
|
|
|
|
i |
|
|
|
|
0 |
скорость ri стремится к rравн – ско- |
||||||
и r ri(ASRT ) . При A SRT |
|||||||||||||||||
рости в равновесии. |
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Сродство всех стадий в цепи последовательных реакций положительно, а их сумма равна суммарному сродству (см. выше). Таким образом, термодинамическое сопряжение (протекание термодинамически невыгодной реакции за счет другой, выгодной) невозможно среди последовательных стадий в стационаром состоянии.
Если есть хотя бы одна стадия с отрицательным сродством, реакция останавливается.
Однако возможно так называемое кинетическое сопряжение в цепных или каталитических реакциях в том случае, когда концентрация промежуточных веществ выше равновесной, если они реагенты (в левой части уравнения), или ниже равновесной, если они продукты. Кинетическое сопряжение возможно при некотором соотношении стадий.
СогласноМ. Будару, приразложенииаммиака2NH3 N2 + 3H2 на пленках W и Mo при 1000 К и давлении 10–4 Па по Оже-спектрам было найдено, что стационарное покрытие поверхности атомами азота в условиях катализа значительно превосходит равновесную концентрацию (т. е. в отсутствии катализа).
Механизм: k1 М– атомметалла
2NH3 2M 2M # N 3H2,
на поверхности; k2 k#1 .
2M # N k 2M N2
Здесь стационарные значения#2 концентраций атомов азота на поверхности значительно больше концентрации, которая должна быть в равновесии с [N2]газ. Десорбция азота хотя и эндотермична, но существенно необратима. Кроме того, катализатор не отравляется адсорбцией азота, что было бы в случае равновесной системы. Здесь кинетическое сопряжение осуществляется вследствие получения
78
сверхравновесных концентраций азота на поверхности в стадиях, предшествующих конечной термодинамически неблагоприятной стадии, т. е. за счет взаимодействия с аммиаком. Соответственно концентрация азота не появляется в уравнении скорости реакции.
Таким образом, кинетическое сопряжение смещает равновесие термодинамически неблагоприятных стадий как в катализе, так и в цепных реакциях.
Можно представить себе, что перед теорией хемосорбции и катализа стоит задача измерения и расчета скоростей обмена энергией между адсорбированными молекулами и твердым телом. Количественныехарактеристикиэтихпроцессовдаютвозможностьустановить механизм физической и химической адсорбции и, следовательно, механизм элементарных актов в катализе. Необходимо изучить влияние различных уровней возбуждения на адсорбцию и катализ
ирешить обратную задачу – о распределении энергии в продуктах десорбции и каталитической реакции. Важнейшей задачей является изучение «возбуждения» самого катализатора (его перестройки и активации под влиянием хемосорбции и каталитической реакции), отклонений от равновесия на макроуровне и образования диссипативных структур.
Равновесные условия, т. е. условия, в которых поступательные
ивнутренние степени свободы реагентов находятся в состоянии термодинамического равновесия, не дают возможности ответить на вопрос, какие формы энергии наиболее эффективны с точки зрения преодоления потенциального барьера и, следовательно, ускорения реакции. Необходимо проводить неравновесные исследования.
6.3.Пучковые исследования
1.Молекулярные пучки – метод исследования обмена энергией между молекулами и поверхностью, а также выяснения элементарных стадий хемосорбции и катализа. Поток молекул реагента, пропущенный из камеры высокого давления через малое отверстие в вакуумную камеру, ударяется о мишень-катализатор. Энергия молекул в пучке определяется устройством источника, размер отверстия источника должен быть меньше среднего свободного пробега молекул. В пучке соударения молекул между собой и со стенкой невоз-
79
можны, отпадают и диффузионные затруднения. Метод позволяет определить: 1) минимальное время жизни молекулы на поверхности, которое ведет к реакции; 2) вероятность реакции или адсорбции при одном столкновении с поверхностью (коэффициент прилипания); 3) зависимость вероятности адсорбции и реакции от энергии молекул в пучке и от температуры поверхности; 4) вероятность дезактивации возбужденных молекул; 5) распределение энергии (поступательной, колебательной и вращательной) в продуктах реакции или рассеянном пучке; 6) угловое распределение десорбированных и рассеянных молекул. Можно найти также зависимость скорости реакции от покрытия поверхности дозированным количеством адсорбента.
2. Ионные пучки. Методики экспериментов определяются характеромрешаемыхзадач. Можно выделить следующие: 1) спектроскопия поверхности твердого тела; 2) плазменная обработка поверхности твердого тела; 3) гетерогенная плазмохимия; 4) поиски новых методов контролируемого изменения кинетики гетерогенных процессов; 5) изучение элементарных актов взаимодействия атомных и молекулярных частиц с поверхностью твердого тела.
Можно использовать низкоэнергетические ионы. Варьируя параметры ионного пучка – знак заряда, электронное состояние, распределение по колебательным уровням, энергию и угол падения, – можно направленно изменять параметры молекул, получающихся при нейтрализации ионов и вступающих на поверхности в различныехимическиепревращения. Можноуказыватьусловия, вкоторых образующиеся молекулы будут преимущественно иметь либо электронное, либо колебательное, либо только поступательное возбуждение. Появиласьвозможностьизучениявкладаразличныхстепеней свободы газовых реагентов в эффект активации.
6.4. Лазерные методы
Благодаря высокой монохроматичности и большой мощности с помощью лазеров можно: 1) обеспечить независимое возбуждение степенейсвободыреагентов, чтобывестиреакциювнужномнаправлении; 2) с высокой чувствительностью определить и идентифицировать промежуточные продукты реакции, измерить энергетическое
80