

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
14 Sequence-Specific
Manipulation of DNA
EXPLOITING THE SPECIFICITY OF BASE-BASE RECOGNITION
In this chapter various methods will be described that take advantage of the specific recognition of DNA sequences to allow analytical or preparative procedures to be carried
out on a selected fraction of a complex DNA sample. For example, one can design chemical or enzymatic schemes to cut at extremely specific DNA sites, to purify specific DNA
sequences, or to isolate selected classes of DNA sequences. Methods have |
been devel- |
|
oped that allow the presence of repeated DNA sequences in genomes to be used as pow- |
||
erful analytical tools instead of serving as roadblocks for mapping and DNA sequencing. |
||
Other methods have been developed that allow the isolation of DNAs that recognize spe- |
||
cific ligands. Finally a large number of programs are |
underway to explore the |
direct use |
of DNA or RNA sequences as potential drugs. |
|
|
In almost all of the objectives just outlined, |
a fundamental strategic |
decision must be |
made at the outset. If the DNA target of interest can be melted without introducing unwanted complications, then the single-stranded DNA sequence can be read, directly, and
the full power of PCR can usually be brought to bear to assist in the manipulation of the DNA target. PCR has been well described in Chapter 4, and there is no need to re-intro- duce the principles here. In those cases where it is not safe or desirable to melt the DNA, alternative methods are needed. Such cases include working with very large DNA, which
will break if melted, and working in vivo. Here a very attractive approach is to use DNA triplexes that are capable of recognizing the specific sequence of selected portions of an
intact |
duplex |
DNA. Triplexes |
may not have been encountered by |
some |
readers before, |
and so their basic properties will be described before their utility is demonstrated. |
|||||
STRUCTURE OF TRIPLE-STRANDED DNA |
|
|
|||
Unanticipated formation of triple-stranded DNA helices was a |
scourge of early experi- |
||||
ments with model DNA polymers. Most of the first available synthetic DNAs were ho- |
|||||
mopolymers like poly dA and poly dT. Contamination of samples or buffers with magne- |
|||||
sium |
ion was |
rampant. DNAs |
love to form triplexes under |
these |
conditions, if the |
sequence permits it. Many homopolymeric or simple repeating sequences can form triplestranded complexes consisting of two purine-rich and one pyrimidine-rich strand or one purine-rich and two pyrimidine-rich strands, depending on the conditions. This is true for DNAs, RNAs, or DNA-RNA mixtures. Eventually conditions were found where the un-
wanted |
formation |
of |
these |
triplexes could be suppressed. The whole issue was forgotten |
and lay |
dormant |
for |
more |
than a decade. Triplexes were rediscovered, under much more |
470

STRUCTURE OF TRIPLE-STRANDED DNA |
471 |
Figure 14.1 Appearance of S1 nuclease hypersensitive sites upstream from the start of transcription of some genes.
interesting circumstances when a decade ago investigators began to explore the chromatin structure surrounding active genes.
The key observation that led to a renaissance of interest in triplexes is a phenomenon called S1 hypersensitivity. S1 nuclease is an enzyme that cleaves single-stranded DNA specifically, usually at slightly acidic pH. It will not cleave double strands; it will not even cleave a single-base mismatch efficiently, although it will cut at larger mismatches. Investigators were using various nucleases to examine the accessibility of DNA segments
near or in genes as a function of the potential for gene expression in particular tissues. Unexpectedly, many genes showed occasional sites where S1 could nick one of the DNA
strands, upstream from the start of transcription, quite efficiently (Fig. 14.1). The phenomenon was termed S1 hypersensitivity. Its implication was that some unusual structure
must exist in the region, rendering the normal duplex DNA susceptible to attack. To iden-
tify |
the sequences responsible for S1 hypersensitivity, upstream sequences were cloned |
||||
and |
tested |
for |
S1 sensitivity. Fortunately they were initially tested within |
the plasmids |
|
used |
for cloning. These |
plasmids were highly supercoiled, and S1 hypersensitive sites |
|||
were |
found |
and |
rapidly |
localized to complex homopurine stretches like the |
example |
shown in Figure 14.2. The S1 nicks were found to lie predominantly on the purine-rich strand. It was soon realized that the S1 hypersensitivity, under the conditions used, required a supercoiled target. The effect was lost when the plasmid was linearized, even by
cuts far away from the purine block.
The problem that remained was to identify the nature of the altered DNA structure responsible for S1 hypersensitivity. The dependence of cleavage on a high degree of superhelicity implied that the sites must, overall, be unwound relative to the normal B DNA duplex. Obvious possibilities were melted loops, left-hand helix formation, or cruciform extrusion (formation of an intramolecular junction of four duplexes like the Holliday structure illustrated in Chapter 1). None of these, however, were consistent with the par-
ticular DNA sequences that |
formed the S1 hypersensitive sites, and |
none could |
explain |
||
why only the purine strand suffered extensive nicking. The key |
observation |
that resolved |
|||
this dilemma was made by |
Maxim Frank-Kamenetskii, then |
working |
in |
Moscow. |
He |
noted that there was a direct correlation between the amount of supercoiling needed to reveal the S1 hypersensitivity and the pH used for the S1 treatment. A quantitative analysis of this effect indicated that both the amount of unwinding that occurred when the S1 hypersensitive site was created, and the number of protons that had to be bound during this
Figure 14.2 DNA sequence of a typical S1 hypersensitive site.
472 SEQUENCE-SPECIFIC MANIPULATION OF DNA
Figure 14.3 |
|
Formation of a DNA |
triplex by disproportionation of two homopurine-homopyrimi- |
|
|
|||||||||||
dine duplexes. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
process could be explained by a simple model, which involved the formation of a specific |
|
|
|
|||||||||||||
intramolecular pyrimidine-purine-pyrimidine (YRY) triple helix. |
|
|
|
|
|
|
|
|||||||||
It is easiest to examine intermolecular triplex formation before considering the ways in |
|
|
||||||||||||||
which such structures might be formed |
intramolecularly at the S1 hypersensitive site. |
|
||||||||||||||
Figure 14.3 illustrates a disproportionation reaction between two duplexes that results in a |
|
|||||||||||||||
triplex and a free single strand. This is precisely the sort of reaction that occurred so fre- |
|
|||||||||||||||
quently in early studies with DNA homopolymers |
and led to the |
presence |
of unwanted |
|
|
|
||||||||||
DNA triplexes. |
If |
such a reaction |
is |
assayed |
|
by |
S1 |
sensitivity, |
disproportionation |
by |
|
|||||
the appearance of a single-stranded polypurine would be detected. The corresponding |
|
|
||||||||||||||
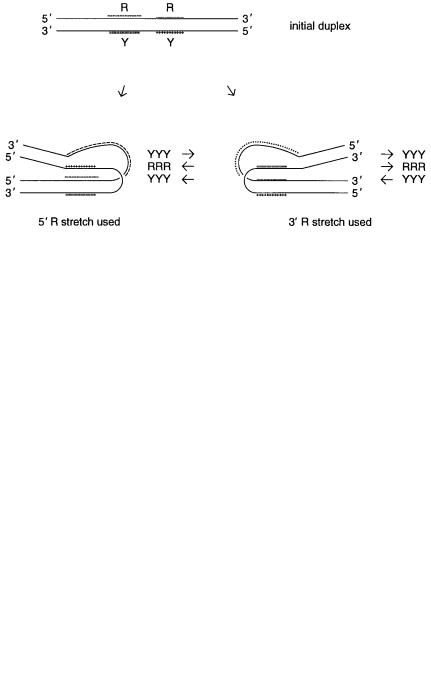
possible intramolecular reactions are illustrated in Figure 14.4. Here a block of homopy- |
|
|||||||||||||||
rimidine sequence folds back on itself (spaced by a short hairpin) to make an intramolec- |
|
|
||||||||||||||
ular triplex; the remaining homopurine |
stretch, not involved in the triplex, is |
left |
as a |
|
||||||||||||
large single-stranded loop. It is |
this loop |
that |
is |
the |
target |
for |
the |
S1 |
nuclease. The |
|
||||||
net topological effect is an unwinding |
of roughly half of the homopurine-homopyrimi- |
|
|
|||||||||||||
dine duplex stretch. This is consistent with what is seen experimentally. In order to form |
|
|
||||||||||||||
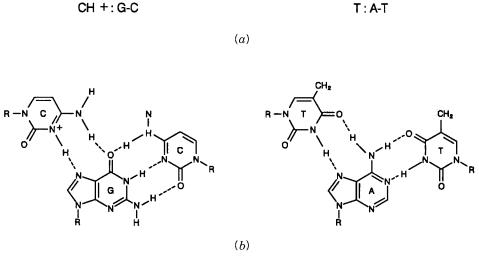
base triplets between two pyrimidines |
and one |
purine, |
a T: A – T |
|
complex can |
form |
|
|
||||||||
directly, but |
a CH |
: G – C |
complex |
requires |
protonation |
of |
the |
N |
|
3 |
of one C, as shown |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
in Figure 14.5.
Figure 14.4 |
Two intramolecular routes for formation of triplexes from a long homopurine- |
|||
homopyrimidine duplex. The structure shown on the right is the one consistant with a large body of |
||||
available chemical |
modification data. |
Y and |
R |
refer, respectively, to homopyrimidine and homo- |
purine tracts. |
|
|
|
|
STRUCTURE OF TRIPLE-STRANDED DNA |
473 |
Figure 14.5 |
Acid-stabilized triplex base pairing schemes. A dash (-) indicates the normal Watson- |
|
Crick base pairing scheme while a colon (:) indicates the base pairs which involve the third strand. |
||
(a) As written schematically. |
(b) Actual proposed structures. |
|
There are two possible isomeric models consistent with the unwinding and pH depen-
dence of formation of the YRY triple strand. In both the two pyrimidine strands run an-
tiparallel to each other; the Watson – Crick pyrimidine strand is antiparallel to the |
purine |
strand; the triplex pyrimidine strand is parallel to the purine strand. The specific structural |
|
models proposed require that the pyrimidine sequences have mirror symmetry. This can |
|
be tested by manipulating particular DNA sequences, and it turns out to be valid. In addi- |
|
tion the two models in Figure 14.4 can be evaluated by looking at the pattern of accessi- |
|
bility of the S1 hypersensitive structure to various agents that chemically modify DNA in |
|
a structure-dependent manner. These studies reveal that the correct model for the S1 hy- |
|
persensitive structure is the one shown on the right in Figure 14.4, where the 3 |
segment |
of the purine stretch is the one incorporated into the triplex. The reason why this structure |
|
predominates is not known.
With the principles of triplex formation in S1 hypersensitive sites understood, a number of research groups began to explore the properties of simple linear triplexes more sys-
tematically. The |
need for superhelical density to drive the formation of triplex can |
be |
avoided simply by working at a low enough pH. In practice, pH 5 to 6 suffices for most |
|
|
sequences capable of forming triplexes at all. A surprise was the remarkable stability of |
|
|
triplexes. They |
can survive electrophoresis, even with lengths as small as 12. Proof |
that |
the third strand lies in the major groove of the Watson – Crick duplex, and that the third,
pyrimidine, |
strand |
is antiparallel to the Watson – Crick pyridine strand, was obtained by |
an elegant |
series |
of experiments in which agents were attached to the ends of the third |
strand that |
were capable of chemically nicking bases on the duplex. The specific pattern |
|
of nicks provided a detailed picture of the structure of the complex (Fig. 14.6). |
||
A second type |
of DNA triplex, stable at pH 7 was soon rediscovered. This purine- |
|
purine-pyrimidine |
(RRY) was precisely the form known two decades before, stabilized |
|

474 SEQUENCE-SPECIFIC MANIPULATION OF DNA
Figure 14.6 |
Structure of DNA triplexes determined from |
chemical |
modification |
experiments. |
(a) |
||
The |
third strand lies in the major groove of the duplex helix. |
|
|
|
(b) Strand directions in structures with |
|
|
two |
pyrimidine strands and one purine strand. |
|
(c) Strand directions in structures with two purine |
|
|||
strands and one pyrimidine strand. Shading in |
(b) |
and |
(c) |
indicates the Watson-Crick duplex. |
|
||
by Mg |
2 or other |
polyvalent cations. This structure could also lead to an S1 hypersensi- |
|||||||||
tive site in supercoiled plasmids. However, here the pyrimidine-rich strand was nicked by |
|
|
|||||||||
the |
enzyme instead |
of |
the purine-rich strand. Some DNA sequences can actually form |
|
|||||||
both types of triplexes, depending on the conditions. The structure of the S1 hypersensi- |
|||||||||||
tive |
sites favored by divalent ions is shown in Figure |
14.7. This particular |
isomer |
is |
the |
||||||
one |
consistent |
with |
the |
observed pattern of modification with various chemical |
agents |
|
|
||||
that react with DNA covalently. Three types of base triples can be accommodated in this |
|
|
|||||||||
structure: G: G – C, A: A – T, and T: A – T. Their patterns of |
hydrogen bonding |
are shown |
|||||||||
in Figure |
14.8. The two non – Watson – Crick base-paired strands |
in |
these |
complexes |
are |
||||||
antiparallel; this is supported by studies on particular DNA sequences. As in the type of |
|
||||||||||
triplex described earlier, the third strand, in this case an additional purine or an additional |
|||||||||||
pyrimidine |
strand, |
lies |
in the major groove of the |
Watson – Crick |
duplex |
(Fig. |
14.6). |
||||
Studies using |
circular |
oligonucleotides can help confirm assignments about the direction |
|
||||||||
of strands in triplexes (Kool, 1995). |
|
|
|
|
|
|
|||||
|
More |
complex triple helices can also be made. An example is shown |
in Figure 14.9. |
||||||||
Here |
all |
three |
strands |
must contain blocks of alternating homopurine and homopyrimi- |
|
|
|||||
dine sequences. The third strand lies down in the major |
groove of the Watson – Crick du- |
|
|||||||||
plex, and alternate blocks made triplexes with two pyrimidine and one purine strand and |
|
|
|||||||||
triplexes with one pyrimidine and two purine strands. As our knowledge of triplex struc- |
|
||||||||||
tures increases, and as base analogs are tested, it will undoubtedly be possible to design a |
|||||||||||
wealth of |
triplexes |
in |
which a third strand can be used to recognize |
a wide variety |
of |
||||||
DNA duplex sequences. Based on experience to date, these triplexes are likely to be quite |
|
||||||||||
stable. One strong caveat to using them in various biological applications must be noted. |
|||||||||||
The |
kinetics of triplex |
formation and dissociation are |
very slow, much |
slower |
than the |
|
|||||
rates of corresponding processes in duplexes.
Figure 14.7 |
Intramolecular triplex structure formed at neutral pH in the presence of Mg |
2 ions. |

Figur e 14.8 |
T riple x base pairing schemes f |
a v o red by Mg |
2 ions. |
(a) As written schematically |
. (b) Actual proposed structures. |
475

476 SEQUENCE-SPECIFIC MANIPULATION OF DNA
Figure 14.9 An example of a more complex triplex structure formed by alternating blocks of purines and pyrimidines. The third strand lies in the major groove of the Watson-Crick duplex. Its interactions with the duplex are indicated by dots.
TRIPLEX-MEDIATED DNA CLEAVAGE
The first application of triplexes to be discussed is their use in recognizing particular duplexes and rendering these susceptible to specific chemical or enzymatic cleavage. This potential was already described briefly in the previous section when chemical derivatives
of the third strand were used |
to help analyze the structure |
of the |
triplex. The appeal of |
||||
this approach is that it will be relatively easy to find or introduce a unique DNA sequence |
|||||||
capable of forming triple strands into a target of interest. Subsequent cleavage at this se- |
|||||||
quence would represent the sort of cut that is extremely useful for executing any of the |
|||||||
Smith-Birnstiel-like mapping strategies we described in Chapter 8. |
|
|
|||||
Chemical cleavage agents that have been tried include Cu-phenanthroline complexes, |
|||||||
iron-EDTA-ethidium bromide complexes, and others shown elsewhere in the chapter. The |
|||||||
types of reactions one would like to be able to carry out with these modified oligonu- |
|||||||
cleotides |
are shown schematically in Figure 14.10. Rather good yields and specificities |
||||||
have |
been |
observed when the chemical cleavage is |
used to cut the complementary strand |
||||
of a duplex (Fig. 14.10 |
a ). Much less success has been had with direct triplex-mediated |
||||||
cleavage |
of a duplex (Fig. 14.10 |
b ). Generally, nicking of one strand of the duplex pro- |
|||||
ceeds very well, but it is difficult to make the second cut needed to affect a true double- |
|||||||
strand cleavage. The reason for this is that many of the chemical agents used are stoichio- |
|||||||
metric rather than catalytic. They have to be reactivated or replaced by a fresh reagent in |
|||||||
order |
to |
be able to perform a second strand cleavage. While elegant |
chemical |
methods |
|||
have been proposed to circumvent this problem, |
to date, specific efficient duplex chemi- |
||||||
cal cleavage has been an elusive goal. However, this has not proved to be a serious road- |
|||||||
block, because alternative methods for using |
triplexes to |
promote |
specific |
enzymatic |
|||
cleavage of duplexes have been very successful. |
|
|
|
|
|||
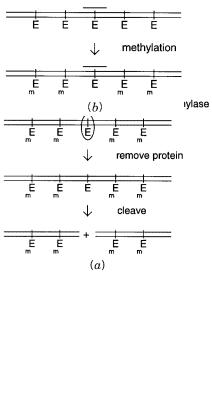
Achilles’s heel strategies are based on the general notion of using restriction methyl- |
|||||||
ases to protect all except a single or small set of protected recognition sites. This renders |
|||||||
most |
of |
the potential sites in the sample resistant to the conjugate restriction nuclease. |
|||||
Then |
the |
protecting agent is |
removed, and the |
nuclease added. Cleavage only occurs at |
|||
Figure 14.10 Triplex-mediated DNA cleavage using a DNA strand containing a chemically reac-
tive group |
(x) that generates radicals. |
(a) Cleavage of a single strand. |
(b) Cleavage of a duplex. |
|
|
|
|
|
|
TRIPLEX-MEDIATED DNA CLEAVAGE |
477 |
|
the sites that escaped the initial protection. These strategies were named by one of their |
|
|||||||
developers, Wacslaw Szbalski, at the University of Wisconsin, by analogy to the myth of |
|
|||||||
Achilles. As an infant Achilles was dipped into the river Styx by his mother which ren- |
|
|||||||
dered him immune to all physical harm except for his heel, which was masked from the |
|
|
||||||
effects of the Styx because his mother was holding him there. Two straightforward exam- |
|
|||||||
ples of Achilles’s heel specific cleavage of DNA are shown in Figure 14.11. These ap- |
|
|||||||
proaches are applicable to very large DNA |
or even intact genomic DNA, since they can |
|
|
|||||
be carried out in situ in agarose. In one |
case it is necessary to find a tight binding site for |
|
||||||
a protein that masks a restriction site |
(Fig. |
14.11 |
a ). |
Examples of suitable protein and |
|
|||
binding sites are lac repressor, lambda repressor, |
|
|
E. coli |
lexA protein, or a host of eukary- |
|
|||
otic |
transcription factors, |
particularly |
viral factors |
like |
the NFAT protein. To be |
useful, |
|
|
the site must contain an internal or nearby flanking restriction site. There is no guarantee |
|
|||||||
that such a site will conveniently exist |
in a target of interest. However, given the large |
|
||||||
number of potentially useful restriction |
sites, there are many possibilities. If necessary, |
|
||||||
for some applications the desired site can always be designed and introduced. |
|
|
||||||
Instead of proteins, triplexes can |
be used to mask restriction sites (Fig. |
14.11 |
b ). It |
|||||
turns |
out that embedding a |
four-base restriction |
site |
in |
a homopurine-homopyrimidine |
|
||
Figure 14.11 |
Achilles’s heel strategies for specific DNA cleavage. |
(a) Blocking a restriction |
en- |
zyme cleavage site |
E with a DNA binding protein. |
(b) Blocking a restriction enzyme cleavage site |
E |
with a triplex. |
M indicates methylation sites. |
|
|
478 |
|
SEQUENCE-SPECIFIC MANIPULATION OF DNA |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
stretch destabilizes the resulting triplex only slightly. However, triplex formation renders |
|
||||||||||||||||||||||
the site totally unaccessible to the restriction methylase. After the remaining restriction |
|
||||||||||||||||||||||
sites have been methylated, conditions are altered to dissociate the triplex. Then the re- |
|
||||||||||||||||||||||
striction enzyme is added, and cleavage is allowed to occur. This Achilles’s heel approach |
|
||||||||||||||||||||||
works very well, even at the level of single sites in the human genome. However, it still |
|
||||||||||||||||||||||
suffers from the limitation that only a small subset of sequences within a target will be |
|
||||||||||||||||||||||
potential sites of triplex-mediated specific cleavage. |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
A |
generalization of the Achilles’s heel approach is |
possible by the use of the |
|
E. coli |
|||||||||||||||||||
recA protein. Developed by Camerini-Otero and elaborated by Szybalski (Koob et al., |
|
||||||||||||||||||||||
1992), this method has been called recA-assisted restriction endonuclease (RARE) cleav- |
|
||||||||||||||||||||||
age (Fig. 14.12). The method is applicable to genomic DNA because all of the steps can |
|
||||||||||||||||||||||
be carried out in agarose. The recA protein has a number of different activities. One of |
|
||||||||||||||||||||||
these is a cooperative binding to single-stranded DNA, leading to a completely coated |
|
||||||||||||||||||||||
complex containing about one recA monomer for every five |
bases |
(Fig. |
14.12 |
|
|
a ). The |
|||||||||||||||||
coated complex will then interact with double-stranded DNA molecules in a search for |
|
|
|||||||||||||||||||||
sequences homologous to the single strand. In |
|
|
|
|
|
|
|
|
|
E. coli |
this process constitutes one of the |
||||||||||||
early steps that eventually leads to strand invasion and recombination. In the test tube, |
|
||||||||||||||||||||||
without |
accessory |
nucleases, |
the |
reaction stops |
if |
a |
homologous |
duplex |
sequence |
is |
|
||||||||||||
found, |
and the third strand remains complexed to this |
homolog, |
even |
if |
the recA |
protein |
|
|
|||||||||||||||
is subsequently removed (Fig. 14.12 |
|
|
|
|
b ). The mechanism of the sequence search is un- |
||||||||||||||||||
known. Similarly the actual nature of the complexes formed with recA protein present or |
|
|
|||||||||||||||||||||
after |
recA protein removal are still not completely understood despite intense efforts to |
|
|||||||||||||||||||||
study |
these processes because of their importance in basic |
|
|
|
|
|
|
|
|
|
E. coli |
biology. Some sort of |
|||||||||||
triple strand is believed to be involved, although this has never been proven. What is key, |
|
||||||||||||||||||||||
however, |
for |
Achilles’s |
heel |
applications, |
is |
that |
the recA |
protein-mediated |
complex |
|
|||||||||||||
blocks |
the access of restriction methylases to duplex DNA sequences contained within it. |
|
|
||||||||||||||||||||
A |
schematic outline of RARE cleavage is given in |
Figure |
14.12 |
|
|
|
|
c . The technique has |
|||||||||||||||
worked well to cut at two selected sites 200 to 500 kb apart in a target to generate a spe- |
|
||||||||||||||||||||||
cific internal fragment, and generation of a 1.3-Mb telomeric DNA fragment that requires |
|
||||||||||||||||||||||
only a single RARE cleavage has been reported. In practice, it has been more efficient to |
|
||||||||||||||||||||||
use a six-base specific restriction system like rather than the four-base systems used with |
|
||||||||||||||||||||||
other |
Achilles’s heel methods. The reason is that a common |
source |
of |
background |
in |
|
|||||||||||||||||
these approaches is incomplete methylation. This produces a diverse distribution of hemi- |
|
|
|||||||||||||||||||||
methylated sites which are cut, albeit slowly, by the conjugate nuclease. The result is a |
|
||||||||||||||||||||||
significant background of nonspecific cleavage. This background |
|
can be markedly re- |
|
|
|||||||||||||||||||
duced by going to the six-base enzyme, since its sites are 16 times less frequent, on aver- |
|
||||||||||||||||||||||
age. Since recA-mediated cleavage is applicable, in principle, to any selected DNA se- |
|
||||||||||||||||||||||
quence, the rarity of six-base cleavage sites does not pose a particular obstacle. |
|
|
|
|
|||||||||||||||||||
The recA protein-coated single strands can be as short as 15 bases for RARE cleavage, |
|
||||||||||||||||||||||
although |
in |
practice |
targets |
two to four times this length |
|
are |
usually |
employed. |
One |
|
|||||||||||||
makes a trade-off between the increased efficiency and specificity obtained with longer |
|
|
|||||||||||||||||||||
complexes, and the lowered efficiency of their diffusion into agarose-embedded DNA |
|
|
|||||||||||||||||||||
samples. Yields of the desired duplex of 40% to 60% have been reported in early experi- |
|
||||||||||||||||||||||
ments. |
It remains |
to |
be |
seen |
how |
generally |
obtainable |
such high yields will be. The |
|
||||||||||||||
power |
of |
RARE |
cleavage in |
physical mapping |
is |
that |
given |
two |
DNA |
probes |
spaced |
|
|||||||||||
within about 1 Mb, RARE cleavage should provide the DNA between these probes as a unique fragment free from major contamination by the remainder of the genome.
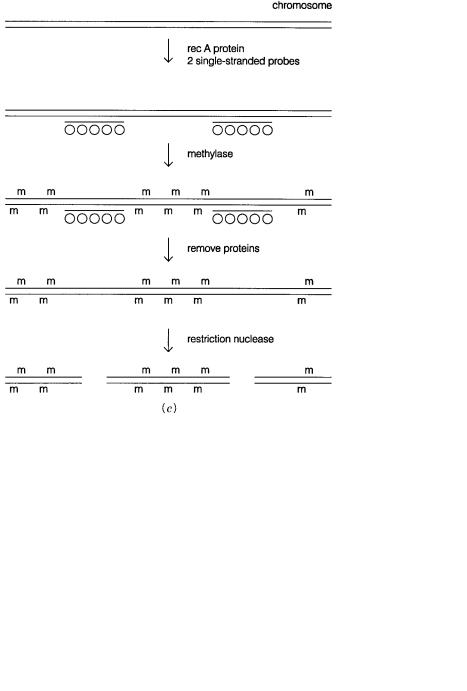
TRIPLEX-MEDIATED DNA CLEAVAGE |
479 |
Figure |
14.12 |
RARE cleavage of |
DNA. |
(a) Complex |
formed between |
E. coli |
recA protein and |
single-stranded DNA. |
(b) Complex between a recA-protein coated DNA strand and the homologous |
|
|
||||
sequence |
in duplex DNA. |
(c) |
Outline |
of the procedure used to |
generate a large DNA fragment |
be- |
|
tween two known sequences by RARE cleavage.