

510 |
SEQUENCE-SPECIFIC MANIPULATION OF DNA |
|
|
|
|
|
|
|||||
been shown to be the case for the intact L1 repeat. Given this general picture, it is not sur- |
|
|||||||||||
prising that certain types of repeats appear to cluster in certain genome regions. While the |
|
|
||||||||||
exact mechanism for this clustering is not understood, a formal mechanism to explain it |
|
|
||||||||||
would simply be to state that the transposition of the mobile elements favors genomic re- |
|
|
||||||||||
gions with certain properties. |
|
|
|
|
|
|
|
|
||||
Alu sequences are preferentially located in Giemsa light, G |
|
|
|
|
C-rich bands. Note that |
|||||||
the Alu’s, |
themselves |
are G |
C |
rich. Their average base composition |
is 56% G |
C, |
||||||
and they have a CpG content that is only 64% of that expected statistically for such a G |
|
|||||||||||
C content. This is remarkable given the overall suppression of CpG throughout most of |
|
|
||||||||||
the genome. Thus Alu’s behave a bit like HTF islands (Chapter 8). It |
is |
not surprising |
|
|||||||||
then, that |
like |
HTF |
islands. Alu’s seem to preferentially associated |
with |
genes. The |
|
||||||
Giemsa light bands are very gene rich, and within many of these genes there are truly re- |
|
|
||||||||||
markable numbers of Alu repeats (in the introns, of course). These Alu’s greatly compli- |
|
|
||||||||||
cate attempts to sequence genomic, gene-rich regions by shotgun strategies (Chapter 10) |
|
|||||||||||
because of the difficulty of assembling the sequence. |
|
|
|
|
|
|
|
|||||
L1 sequences occur preferentially in dark bands. These bands are A |
|
|
|
|
T rich. The L1s |
|||||||
themselves |
are |
A |
|
T rich (58%). They are also extremely deficient in CpG sequences |
|
|||||||
with only 13% of what would be expected statistically. Thus we can conclude from both |
|
|
||||||||||
the patterns of |
Alu’s |
and L1’s that like attracts like, but the mechanism behind |
this |
re- |
|
|||||||
mains unclear. Earlier we indicated that the pattern of Alu distribution was really bipha- |
|
|
||||||||||
sic. Presumably the Alu-rich phase seen in renaturation experiments corresponds to DNA |
|
|
|
|||||||||
from light bands, but this has not been formally proved. |
|
|
|
|
|
|
||||||
The final class of well-characterized interspersed repeats are the VNTRs. These are |
|
|||||||||||
preferentially |
located |
in telomeric light Giemsa bands, although they |
are |
spread |
well |
|
||||||
enough through the genome to be generally useful as genetic markers. The reason why |
|
|
|
|||||||||
VNTRs cluster near the telomeres is unknown. However, it is worth noting that the fre- |
|
|||||||||||
quency of meiotic recombination appears to be very high in the telomeric regions and that |
|
|
|
|||||||||
one mechanism of VNTR growth and shrinkage is recombination. There is no way to tell |
|
|
||||||||||
at present whether any causal relationships existing among these observations. However, |
|
|
|
|||||||||
they represent a tantalizing area for future study. |
|
|
|
|
|
|
|
|||||
PCR BASED ON REPEATING SEQUENCES |
|
|
|
|
|
|
|
|
||||
Sequences in the Alu and L1 families are similar enough so that a single PCR primer can |
|
|||||||||||
be used to initiate DNA synthesis within a large number of these elements. Some of the |
|
|
||||||||||
common Alu primers are summarized in Figure 14.38. These are chosen to try to focus on |
|
|
||||||||||
the most-conserved regions of the repeats within known human sequences without select- |
|
|
|
|||||||||
ing sequences that are also conserved in rodents. Some Alu primers are tagged with ex- |
|
|
||||||||||
tensions to allow more efficient amplification after the first few rounds of PCR where an |
|
|
||||||||||
inexact or very short match between primer and |
target template |
may |
be |
occurring |
|
|
||||||
(Chapter |
4). |
The |
general situation in which these primers are |
applicable is |
shown |
in |
|
|||||
Figure 14.39. Neighboring copies of a repeat can have inverted configurations (head to |
|
|||||||||||
head or tail to tail) or tandem configurations (head to tail). In the former case, a single |
|
|||||||||||
PCR primer will serve to amplify the DNA between the repeats. In the latter case, two |
|
|
||||||||||
primers must be used. Inter-Alu PCR is a very powerful tool because so much of the hu- |
|
|
||||||||||
man genome |
is |
dense in Alu sequences, and Alu |
sequences in humans are |
well |
diverged |
|
|
|
||||

PCR BASED ON REPEATING SEQUENCES |
511 |
Figure 14.38 DNA sequences of some of the primers commonly used to amplify DNA between repeated Alu sequences by PCR.
from those of rodents. Thus a significant fraction of the human genome is potentially am- |
|
|
plifiable by inter-Alu PCR. The L1 |
repeat is less useful in this regard because it |
is rarer |
and because its sequence is more conserved in rodents and human. Despite this limitation |
|
|
inter-L1 PCR or PCR between L1 |
and Alu sequences can still be helpful tools. |
|
It would take most of a chaper to |
describe the myriad applications of inter-Alu or |
in- |
ter-L1 PCR in detail. We will just list a number of the most prominent applications, and then illustrate a few in more detail. Inter-Alu PCR is helpful whenever one needs to selectively amplify the human component in a nonhuman background. Not all the human DNA
will be amplified. In general, one can expect good amplification wherever two Alu sequences with close sequence homology to the primers used lie in the correct orientation
within |
a |
few kb of |
each other. This will be quite frequent in Alu-rich regions of the |
|
genome, much rarer in other regions. For example, inter-Alu PCR will selectively amplify |
||||
the |
human |
component in a rodent hybrid cell line. It will preferentially amplify the YAC |
||
DNA |
in |
a |
background |
of yeast DNA. Combining YAC vector primers and Alu primers |
Figure 14.39 Some of the arrangements of interspersed repeats (arrows) that can be amplified by
PCR using primers selected from the sequence of the repeat.
512 |
SEQUENCE-SPECIFIC MANIPULATION OF DNA |
|
|
|
|
|
|
|||
will preferentially amplify human DNA cloned near the ends of YAC inserts. These are |
|
|||||||||
exactly the samples most desirable as probes for YAC |
walking techniques. Single-sided |
|
||||||||
Alu amplification procedures have also been described for regions where Alu’s are too |
|
|||||||||
dilute to allow efficient inter-Alu PCR (Quereshi et al., 1994). Single-sided Alu PCR is |
||||||||||
also a very useful method to treat chromosome-specific hncDNA libraries, as described in |
|
|||||||||
Chapter 11. |
|
|
|
|
|
|
|
|
|
|
The pattern of bands amplified by inter-Alu PCR from a whole human chromosome is |
|
|||||||||
often too complex to analyze (although inter-L1 PCR is helpful in such applications). |
|
|||||||||
Sometimes, with particular primers and conditions, the number of amplified bands can be |
|
|||||||||
reduced to of the order to 40. In such cases a significant fraction of these bands appears to |
|
|||||||||
be polymorphic in the population, and thus the Alu PCR provides a very convenient and |
|
|||||||||
easily used set of multiplexed genetic markers (Fig. 14.40). |
|
|
|
|||||||
In contrast to most attempts to |
amplify DNA from the whole genome, the pattern of |
|
||||||||
bands from a few Mb of DNA is |
usually quite clear and |
diagnostic. Thus inter-Alu |
PCR |
|
||||||
is a powerful fingerprinting method. The added incentive is that the PCR-amplified bands |
|
|||||||||
seen in an electrophoretic analysis can be cut out and used as single-copy hybridization |
|
|||||||||
probes (competing any residual Alu sequences, as needed). Thus inter-Alu PCR finger- |
|
|||||||||
printing has been applied to YACs, |
chromosome fragments, radiation hybrids, and PFG |
|
||||||||
gel slices, and it has been used to isolate single-copy DNA probes from all of these kinds |
|
|||||||||
of samples. The products of inter-Alu PCR reactions are very useful for FISH mapping. |
|
|||||||||
They are usually complex and |
concentrated enough to yield good results and allow |
the |
|
|||||||
rough map position of the sample to be identified. Inter-Alu PCR products are also very |
|
|||||||||
useful for cross-connecting libraries. |
|
|
|
|
|
|
|
|||
An example of inter-Alu PCR |
applied to |
the analysis |
of PFG gel |
slices |
is shown in |
|
||||
Figure 14.41. Here a |
Not |
I-digested hybrid cell line containing chromosome 21 as its only |
||||||||
human component was fractionated by PFG under a number of different conditions to |
|
|||||||||
maximize the resolution of indivdiual size regions. Each gel lane was sliced into 40 frag- |
|
|||||||||
ments, and each fragment was subjected to inter-Alu PCR. In most cases lanes showed |
|
|||||||||
discrete patterns of several amplified bands, and adjacent slices often showed quite differ- |
|
|||||||||
ent patterns (Fig. 14.42). Thus the carryover |
of material from slice to slice during |
the |
||||||||
PFG and subsequent steps is not so |
serious as to obscure the fractionation. This means |
|
||||||||
that individual PCR products from analytical gels like the one shown in Figure 14.42 can |
|
|||||||||
be cut out, reamplified, and used as |
immortal specific single-copy DNA probes. Even if |
|
||||||||
the PFG gel slice contained more than one genomic human |
|
|
|
Not |
I fragment, the individual |
|||||
PCR products from that slice are each likely to derive from only a single genomic frag- |
|
|||||||||
ment. Thus they constitute a very |
convenient |
source |
of |
new |
single-copy |
human |
DNA |
|
||
probes. |
|
|
|
|
|
|
|
|
|
|
The method illustrated in Figures 14.41 and 14.42 is very helpful in the later stages of |
||||||||||
physical mapping where most fragments of a chromosome are located and the goal is to |
|
|||||||||
obtain new probes for unassigned bands as efficiently as possible. One problem with the |
|
|||||||||
kind of results shown in Figure 14.42 is that the number of new probes provided by a sin- |
|
|||||||||
gle experiment is very large, frequently a hundred or more. Before selecting probes for |
|
|||||||||
further study, usually one would like to know something about their regional location on |
|
|||||||||
the chromosome of interest. The standard way to do this is to take a probe of interest and |
|
|||||||||
hybridize it to a mapping panel of chromosome deletions as we described in Chapter 8. |
|
|||||||||
However, this is far too inefficient |
when a hundred or |
more probes must be mapped at |
|
|||||||
once. An alternative approach, useful |
for YACs or slices of PFG fractionations, |
is shown |
|
|||||||
in Figure |
14.43. Ideally |
what one would like to |
do |
is take DNA from |
hybrid |
cell lines |
|
|||
PCR BASED ON REPEATING SEQUENCES |
513 |
|
|
|
|
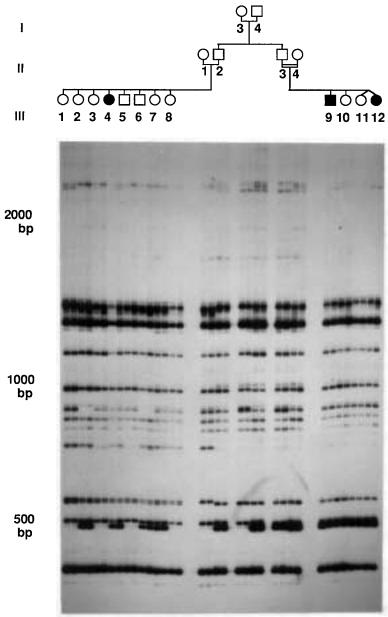
Figure 14.40 |
An example of Alumorphs: Polymorphic genomic DNA sequences amplified by in- |
||||
ter-Alu PCR. Analysis of two |
pseudo – vitamin D-deficient rickets (PDDR) families (affected indi- |
||||
viduals are |
indicated by |
filled |
symbols). The |
32 |
of PCR amplification using an |
P-labeled products |
|||||
Alu-specific |
primer were |
analyzed by electrophoresis |
in nondenaturing 6% polyacrylamide |
gel. |
|
Each individual from the pedigree shown on the top of the autoradiogram was analyzed in duplicate
by two independent PCR amplifications, shown in two adjacent lanes on the gel. Molecular size markers are indicated at left. Taken from Zietkiewicz et al. (1992).

Figure 14.41 Schematic example of the use of inter-Alu PCR to preferentially amplify human DNA from PFG-fractionated large restriction fragments in a hybrid cell line.
Figure 14.42 Inter-Alu PCR products from 10 consecutive slices of a PFG-fractionated Not I digest of a hybrid cell line containing chromosome 21 as its only human component. From Wang et
al. (1995).
PCR BASED ON REPEATING SEQUENCES |
515 |
|
|
|
|
|
|
|
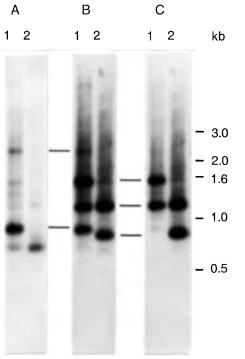
Figure 14.43 |
Example of how inter-Alu PCR products can be assigned, en masse, to chromosome |
|
|
regions. Inter-Alu probes generated from DNA from cell lines 8q |
|
|
|
|
(A), R2-10W (B), or 21q (C) |
||
were used to assign inter-Alu gel slice products regionally. Conventional gel lanes (lanes 1 and 2) |
|
||
containing inter-Alu products generated from template DNA contained in two different PFG slices |
|
|
|
are shown. Note that each cell line specific probe is hybridized to a different set of inter-Alu PCR |
|
||
products. From Wang et al. (1995). |
|
|
|
containing human chromosome fragments and use this as a hybridization probe against a
blot of a gel-fractionation of all the PCR products from a set of YACs or Not I fragments. The difficulty is that the complexity of the source DNA is too large, and one needs a way
to reduce it and label the human component selectively in order to obtain efficient hybridization. However, inter-Alu PCR products from these cell lines provide the precise DNA subpopulation needed for efficient hybridization to inter-Alu PCR products from YACs or PFG fractions. By selectively amplifying the same segments of the DNA in both
the hybridization probe and the hybridization target, one achieves enormously rapid and specific hybridizations. This same principle can be applied whenever inter-Alu PCR products are used for fingerprinting or for cross-connecting libraries.
The pattern of PCR products between repeating sequences can also be used to provide information about the distribution of repeats in the genome. With Alu, the patterns are too
complex to analyze on a whole genome level. The situation is much more favorable with |
|
the 5 L1 sequence. To estimate the number of PCR products expected with a single 5 |
L1 |
primer, Yoshiyuki Sakaki assumed that there were about 3000 copies of the L1 repeat or |
|
one per Mb. This estimate is on the low side of the range reported by others (Table 14.3). |
|
Suppose that the PCR range is 2 kb. Only a quarter of the L1’s within this range will be |
|
516 SEQUENCE-SPECIFIC MANIPULATION OF DNA
oriented head to head and thus amplified by the primer. So |
the |
expected |
frequency |
of |
|
|||||||||||||||||
PCR products per genome can be estimated as |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Number |
|
(3 |
3 |
|
|
|
|
|
6 |
|
|
|
|
|
|
|
3 |
per 1.5 |
||||
|
10 |
Spacing) |
|
|
(10 |
) Range (2 |
10 ) Orientation (0.25) |
|||||||||||||||
genome |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In actuality about 20 genomic |
PCR products are seen. This presumably indicates that |
|
||||||||||||||||||||
L1’s are clustered, which is in |
accord with observations we have described previously. |
|
||||||||||||||||||||
Note, however, that if we took a higher estimate for the number of 5 |
|
|
|
|
L1 sequences, say |
|||||||||||||||||
1.5 |
104 copies, then |
the |
expected number |
of products |
would be |
7.5 |
per |
genome, and |
|
|||||||||||||
the evidence for clustering, from this one experimental result alone, would be much less |
|
|||||||||||||||||||||
compelling. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
PCR amplification schemes can also be based on tandemly repeating dinucleotide or |
|
||||||||||||||||||||
trinucleotide sequences. These are too infrequent to allow amplification between repeats. |
|
|||||||||||||||||||||
Instead, single-sided amplification methods are used. Alternatively, repeating-sequence- |
|
|||||||||||||||||||||
containing fragments are captured by |
hybridization with an |
immobilized |
single-strand, |
|
||||||||||||||||||
and then the released repeats are amplified in a number of different ways. These proce- |
|
|||||||||||||||||||||
dures are quite efficient (Broude et al., 1997; Kandpal et al., 1994). |
|
|
|
|||||||||||||||||||
REPEAT |
EXPANSION |
DETECTION |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
A final example of the use of DNA |
amplification based on interspersed repeating se- |
|
||||||||||||||||||||
quences is shown in Figure 14.44. This illustrates a newly developed technique called re- |
|
|||||||||||||||||||||
peat expansion detection (RED) which is designed as a way to specifically isolate very |
|
|||||||||||||||||||||
large tandemly repeating DNA sequences such as the expanded triplet repeats found in |
|
|||||||||||||||||||||
fragile X syndrome and other human disease alleles (Chapter 13). RED uses the ligase |
|
|||||||||||||||||||||
chain reaction (Chapter 4) instead of PCR. Oligonucleotide probes consisting of 11 to 17 |
|
|||||||||||||||||||||
tandem triplet repeats are annealed to target DNA in the presence of a thermostable DNA |
|
|
||||||||||||||||||||
ligase. Repeated cycles of denaturation and renaturation are carried out. Long repeated |
|
|||||||||||||||||||||
triplet alleles in the target will promote more effective ligation of the probes than short |
|
|||||||||||||||||||||
alleles, and |
a |
more |
complex |
set |
of |
ligated |
products |
will |
result. |
This |
can |
be |
detected |
|
||||||||
by electrophoresis after hybridization with the complementary triplet repeating sequence. |
|
|||||||||||||||||||||
This |
method is a |
very promising approach to the |
discovery |
of |
new |
genes, |
where |
unsta- |
|
|||||||||||||
ble triplet repeats may be responsible for producing disease alleles. Other methods for |
|
|||||||||||||||||||||
repeat |
expansion |
detection |
may |
be |
based on the observation that PCR amplification |
|
||||||||||||||||
of many |
long |
triplet |
repeats |
is |
inefficient |
at |
best |
and |
often |
fails |
completely |
(Broude |
|
|||||||||
et al., 1997).
Figure 14.44 Schematic illustration of the repeat expansion detection (RED) procedure used to identify cells with large, potentially disease-causing repeated triplet alleles. Adapted from Schalling
et al. (1993).

APTAMER SELECTION STRATEGIES |
517 |
APTAMER SELECTION STRATEGIES |
|
|
|
|
|
|
A relatively recently developed set of strategies combines |
physical purification and |
PCR |
|
|||
to select DNAs (or RNAs or proteins) with |
desired |
sequences |
or |
binding |
properties. |
|
These methods appear to be powerful enough that in some cases one can start with all 4 |
n |
|||||
|
||||||
possible nucleic acids of length |
n and |
find the one |
or |
few with |
the optimal affinity for a |
|
given target. Among the potential applications are: |
|
|
|
|
|
|
•Purification of DNA sequences with the highest affinity for a given protein, ligand, or drug (such molecules have been termed aptamers)
•Purification of RNA sequences with the highest affinity for a given DNA (via duplex or triplex), RNA, protein, ligand, or drug
•Purification of protein sequences with the highest affinity for a given receptor, ligand, or drug (also called aptamers)
It is relatively easy to make all 4 |
n |
possible DNAs of length |
n just by adding all four |
dpppN’s at each step in automated DNA synthesis. More restricted mixtures can be made |
|
||
by an obvious extension of this approach. |
|
|
|
The general principle behind select strategies is shown in Figure 14.45. A complex |
|||
mixture is allowed to bind to an immobilized target of interest, usually at fairly low strin- |
|||
gency. Those species that do bind are eluted, and PCR amplification is used to regenerate |
|||
a population of molecules comparable to |
the initial total concentration. Now, however, |
||
this population should be enriched for |
molecules that |
have some affinity for the target. |
|
The cycles of affinity purification and amplification can be repeated as often as needed, |
|||
until the complexity of the mixture becomes small enough to analyze. In the perfect case |
|||
only a single species would remain, and |
if necessary, the stringency of the affinity step |
||
could be progressively increased during successive cycles. In actual cases a mixture of |
|||
molecules will be seen, but this will eventually attain a small enough complexity so that |
|||
individual components can be cloned and examined. Alternatively, a powerful approach is |
|
||
to sequence the mixture of molecules remaining after a large number of cycles of select |
|||
purification. If certain positions within the DNA (or RNA or protein) are required for |
|||
affinity, and others are not, the sequence of the mixture will show conserved residues |
at |
||
some positions, which will be unambiguously identified, and mixtures of residues (usually refractory to analysis) at other positions.
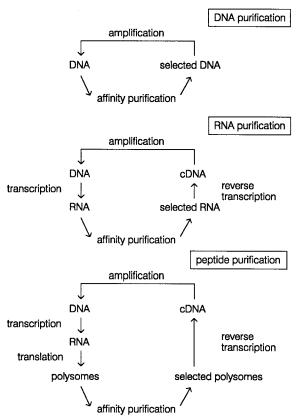
For the select approach to work, one needs a fairly good affinity purification with relatively little nonspecific background. Three potential implementations of the select strategy are shown in Figure 14.46. These allow for purification of DNAs, RNAs, or proteins with particular affinity properties. A number of highly successful examples of the applica-
tion of selection strategies are summarized in Table 14.4. Such strategies are also effec-
Figure 14.45 Basic principle behind a select strategy for purification of sequences with specific affinity properties.
518 SEQUENCE-SPECIFIC MANIPULATION OF DNA
Figure 14.46 |
Select strategies that have been proposed for purifying DNAs, and DNAs coding for |
|
|
|||||||
RNAs, and proteins with particular properties |
that can be converted |
to |
differential |
affinities. |
|
|
||||
Adapted from Irvine et al. (1991). |
|
|
|
|
|
|
|
|||
|
TABLE |
14.4 High-Affinity Aptamers |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Target |
|
Library |
Nucleotides |
a |
Rounds |
b |
Motif |
K d (nM) |
|
|
|
|
|
|||||||
|
E. coli |
rho factor |
RNA |
30 |
|
8 |
|
Hairpin |
|
1 |
|
E. coli |
metJ protein |
RNA |
40 |
|
15 |
|
Unknown |
|
1 |
|
HIV-1 rev protein |
RNA |
32 |
|
10 |
|
Bulge |
|
1 |
|
|
sPLA2 |
RNA |
30 |
|
12 |
|
Complex |
|
1 |
|
|
|
|
Modified |
30 |
|
10 |
|
Pseudoknot |
|
1 |
|
|
|
RNA |
|
|
|
|
|
|
|
|
Basic fibroblast |
RNA |
30 |
|
13 |
|
Hairpin |
|
0.20 |
|
|
growth factor |
|
|
|
|
|
|
|
|
|
|
Vascular endothelial |
RNA |
30 |
13 |
|
Hairpin/bulge |
|
0.20 |
||
|
growth factor |
|
|
|
|
|
|
|
|
|
|
SLE monoclonal |
ssDNA |
40 |
8 |
Unknown |
1 |
|
|||
|
antibody |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Source: Adapted from Gold et al. (1995).
aLength of the random region.
bRounds of selection used.
|
|
|
|
|
|
|
|
|
|
|
APTAMER |
SELECTION STRATEGIES |
519 |
||
tive for finding consensus nucleic acid binding sequences to known proteins such as tran- |
|
|
|||||||||||||
scription factors (Pollack and Treisman, 1990; Nallur, et al., 1996). For proteins the select |
|
||||||||||||||
strategy shown may well be too cumbersome to use in practice. However, as an alterna- |
|
|
|||||||||||||
tive to PCR amplification, one can use in vivo amplification for proteins instead. An ele- |
|
|
|||||||||||||
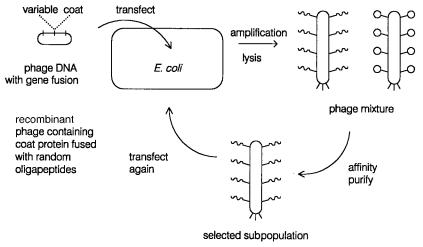
gant way to do this is bacteriophage display, shown schematically in Figure 14.47. Here |
|
|
|||||||||||||
the random coding sequence of interest is subcloned as a fusion with a surface coat pro- |
|
|
|||||||||||||
tein of the bacteriophage M13. Either a minor coat protein is used with only five copies or |
|
|
|||||||||||||
the major coat protein |
with |
thousands |
of copies |
is used. Each bacteriophage plaque |
will |
|
|
||||||||
be a clonal population representing one particular variant. Mixtures of plaques can be |
|
|
|||||||||||||
subjected to cycles of selection by physical affinity to the target of interest, and the suc- |
|
||||||||||||||
cessive |
populations |
of |
bacteriophage that remain will begin |
to |
be |
populated |
more |
and |
|
|
|||||
more with clones with the desired affinity properties. |
|
|
|
|
|
|
|
|
|
||||||
Note that there is no reason why one must start with totally random sequences in select |
|
|
|||||||||||||
or bacteriophage display strategies. In many cases there will be existing structures with |
|
|
|||||||||||||
properties similar to the optimum behavior desired. In this |
case random |
mutagensis of |
|
|
|||||||||||
just a small portion of an existing macromolecule can be used as a starting point try to se- |
|
|
|||||||||||||
lect a more desirable variant. Despite the intrinsic attractiveness of bacteriophage display, |
|
|
|||||||||||||
this approach does have a number of limitations. Only monomeric target proteins can be |
|
|
|||||||||||||
examined by bacteriophage display. The protein targets must |
be |
able |
to |
fold |
properly |
|
|
||||||||
within |
E. coli, |
and they must be oriented in the fusion |
so |
that |
the |
site |
that |
generates their |
|
||||||
affinity for the ligand or target is accessible. |
|
|
|
|
|
|
|
|
|
|
|||||
A generalization of select strategies has been proposed |
by |
Sydney |
Brenner |
and |
|
||||||||||
Richard Lerner; it is called encoded combinatorial chemistry. The basic idea involves tag- |
|
||||||||||||||
ging linear oligomers of any type of residue, with a PCR-amplifiable specific DNA se- |
|
|
|||||||||||||
quence that is a unique identifier of the particular oligomer. The general chemical struc- |
|
|
|||||||||||||
ture needed is shown in Figure 14.48. Here the variable DNA identifier is placed between |
|
|
|||||||||||||
two constant PCR primers, and one of these is attached via a hub to the oligomer, which |
|
|
|||||||||||||
could be a nucleic acid, a peptide, an oligosaccharide, |
or really any kind of organic |
|
|||||||||||||
species |
that can |
be |
built |
up in a |
stepwise |
fashion. The |
number |
of |
different |
types |
of |
|
|||
Figure 14.47 Bacteriophage display method for purifying DNA coding for proteins with selectable affinity properties.