

480 |
SEQUENCE-SPECIFIC MANIPULATION OF DNA |
|
|
|
|
|
|
|
SEQUENCE-SPECIFIC DNA CAPTURE |
|
|
|
|
|
|
|
|
In the majority of cases, the motivation behind attempts to develop ways for sequence- |
|
|||||||
specific DNA cleavage is to provide access to particular |
|
DNA sequences, as just de- |
|
|||||
scribed above for RARE cleavage. A more direct approach would be to develop methods |
|
|||||||
for the specific capture of DNA sequences of particular interest. There are a number of |
|
|||||||
different approaches that have been used to purify DNAs based on sequence rather than |
|
|||||||
size. Biological methods we have already described include PCR or differential cloning. |
|
|||||||
These are extremely effective, but because the steps are |
usually carried out in solution |
|
||||||
and involve a number of enzymes, the targets of such purifications are usually limited in |
|
|||||||
size. Large insert cloning systems have been described, but these are not yet applicable to |
|
|||||||
differential cloning. For example, YAC cloning can be used |
to immortalize a set |
of large |
|
|||||
DNA fragments, but then this set has to be screened to identify the clone containing a |
|
|||||||
particular sequence of interest. Such screening can be quite time-consuming. Instead of |
|
|||||||
screening, we are concerned here with methods that |
|
select the target of interest. |
||||||
Purification techniques are a natural way to select molecules with desired properties such |
|
|||||||
as specific DNA sequences. |
|
|
|
|
|
|
|
|
|
In principle, a DNA sequence could be selected by cloning, by PCR, by binding to a |
|
||||||
specific protein, by binding to the complementary strand to |
form a duplex, or by binding |
|
||||||
to a third strand to form a triplex. In each of these approaches the desire is a simple se- |
||||||||
quence-specific purification method. In this section we will limit our attention to physical |
|
|||||||
purifications based solely on DNA base interactions. Such |
methods have been called in |
|
||||||
the past, perhaps somewhat inelegantly, DNA fishing. Differential PCR and cloning meth- |
|
|||||||
ods will be described later in the chapter. The advantage of the pure, physical methods is |
|
|||||||
that if successful, they are very easy to implement and easy |
to scale up to large |
numbers |
|
|||||
of samples or large quantities. A key obstacle in the use of |
such methods |
is that |
once the |
|
||||
DNA target is captured by a probe, there either must be an |
efficient way to release it, or |
|
||||||
one has to have an efficient way of working directly with |
the immobilized DNA (as in |
|
||||||
solid state DNA sequencing, described earlier in Chapter 9). |
|
|
|
|
||||
TRIPLEX-MEDIATED DNA CAPTURE |
|
|
|
|
|
|
|
|
The major advantage in using a third strand to capture a |
DNA duplex is that there is no |
|
||||||
need to melt the duplex. This avoids potential damage arising from any preexisting |
nicks |
|
||||||
in the DNA strands. It also helps avoid many of the complications caused by the extensive |
|
|||||||
interspersed repeats in the DNA of higher organisms. There are a number of potential ap- |
|
|||||||
plications for triplex capture of DNA. For example, triplex formation could be used, in |
|
|||||||
principle, to isolate from a library all those clones that contained specific homopurine-ho- |
|
|||||||
mopyrimidine sequences such as (AG) |
|
n , (AAG) |
n , or (AAAG) |
n . These clones are all poten- |
||||
tially useful genetic probes, since all are VNTRs. Such clones have been isolated in the |
||||||||
past by screening libraries by hybridization |
with the repeated seqeunce. However, |
if one |
|
|||||
is |
interested in a large set of such clones, |
each one detected by hybridization |
has to |
be |
||||
picked by hand for future use. It would be far simpler to isolate the entire set of VNTRcontaining clones in a single physical purification step. A survey of simple repeated sequence motifs in the GenBank database several years ago is shown in Table 14.1. This indicates that a significant fraction of all simple sequence VNTRs known to date could be amenable to triplex-mediated capture because they involve homopurine repeats.
|
|
|
|
|
|
|
TRIPLEX-MEDIATED |
DNA |
CAPTURE |
|
481 |
|||
TABLE 14.1 |
Repeating Single Sequence Motifs in the Genbank Database |
|
|
|
|
|
|
|||||||
(1991) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dinucleotide |
|
AG |
|
24 |
|
|
|
|
|
|
|
|||
|
|
AT |
18 |
|
|
|
|
|
|
|
||||
|
|
AC |
86 |
|
|
|
|
|
|
|
||||
|
|
TG |
1 |
|
|
|
|
|
|
|
||||
Trinucleotide |
AAC |
14 |
|
|
|
|
|
|
|
|
||||
|
|
CCG |
12 |
|
|
|
|
|
|
|
||||
|
|
AGG |
|
9 |
|
|
|
|
|
|
|
|||
|
|
AAT |
7 |
|
|
|
|
|
|
|
||||
|
|
AGC |
10 |
|
|
|
|
|
|
|
||||
|
|
AAG |
|
3 |
|
|
|
|
|
|
|
|||
|
|
ATC |
2 |
|
|
|
|
|
|
|
||||
|
|
ACC |
2 |
|
|
|
|
|
|
|
||||
Tetranucleotide |
|
AAAG |
|
20 |
|
AGCC 1 |
|
AACT |
1 |
|
|
|
||
|
|
AAAT |
22 |
AGAT |
9 |
ACAT |
1 |
|
|
|||||
|
|
ACAG |
3 |
ATAG |
3 |
ACGC |
1 |
|
|
|||||
|
|
ACAT |
2 |
AGCG |
1 |
ACTG |
2 |
|
|
|||||
|
|
AAGG |
|
8 |
ATCC |
8 |
|
AGGG 1 |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
AAGG |
|
13 |
AATC |
4 |
AGCT 1 |
|||||||
Lohger |
|
AAAAG |
1 |
|
|
|
|
|
|
|
||||
|
|
AAAAAAAAAG |
|
1 |
|
|
|
|
|
|
|
|||
|
|
TTTTTG |
1 |
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Source: Adapted from results summarized by Lincoln McBride.
Note: Underlined sequences are triplex selectable.
Two other potential |
applications for triplex capture concern the purification of a spe- |
|
cific DNA sequence. If a genomic fragment contains a known specific homopurine se- |
||
quence, |
one should be |
able to capture this sequence away from the entire genome by |
triplex formation on a solid support. Alternatively, such a sequence could be introduced |
||
into a desired section of a genome to allow subsequent capture of DNA from this region. |
||
Such approaches, once they have matured, could potentially expedite the analysis of se- |
||
lected |
megabase regions |
of DNA considerably. The second potential use of triplex cap- |
ture would be to separate clones from host cells. Here the triplex forming sequence would
be built into the vector used for cloning. After cell lysis, triplex capture would be used to retain the cloned DNA and discard the DNA of the host. This has many appealing features. Once host DNA is gone, direct analysis of cloned DNA is possible without the need for radiolabeled DNA probes or specific PCR primers. Host DNA leads to considerable background problems when low-copy number vectors like YACs are used. The sensitivity
of hybridization or PCR analyses would be increased considerably if host DNA could be
removed in advance. |
|
|
|
|
||
A number |
of |
different ways to use triplexes to purify DNA have |
been |
explored. |
||
Although |
much |
more |
development work needs to be done, the preliminary findings |
are |
||
very promising. In our work, magnetic microbeads were used as the solid support. The |
||||||
power of this general approach was described earlier in applications for solid state DNA |
||||||
sequencing. Magnetic microbeads are available commercially that are coated with strep- |
||||||
tavidin. |
Streptavidin |
will bind specifically to virtually any biotinylated |
macromolecule. |
|||
Any desired small |
DNA |
sequence can be prepared with a 5 |
|
-biotin by the use of biotin |
||

482 |
SEQUENCE-SPECIFIC MANIPULATION OF DNA |
|
|
|
phosphoramidite in ordinary automated oligonucleotide |
synthesis. Longer 5 |
|
-biotinylated |
|
probes can be made by PCR using 5 |
-biotinylated primers. Alternatively, internally bi- |
|||
otinylated |
DNA sequences can be made by PCR |
using biotinylated base analogs as |
|
|
dpppN substrates. Some of these compounds are incorporated quite efficiently by DNA |
|
|
||
polymerases. Once attached to the streptavidin microbeads, biotinylated DNA probes are |
|
|||
still quite accessible for hybridization or for triplex formation. A schematic illustration of |
|
|||
the way microbeads have been used to develop a simple triplex capture method of DNA |
|
|
||
purification is shown in Figure 14.13. The key |
point is that a permanent magnet can be |
|
||
used to hold the beads in place while supernatant is removed and exchanged. This allows |
|
|||
very rapid and efficient exchanges of reagents. At pH 5 or 6 any duplex DNA captured by |
|
|||
the complementary sequence on a bead will remain attached. At pH 8 the captured DNA |
|
|
||
will be readily released and removed with the supernatant. |
|
|
||
Three different experiments have been used to |
test the ease and efficiency of |
triplex |
|
|
DNA capture with magnetic microbeads. In the first, |
an artificial mixture was made of |
|
||
two plasmids. One contained just a vector with no known forming capability, which in- |
|
|||
cluded a lacZ gene. When this plasmid is transformed into |
E. coli, |
blue colonies are pro- |
||
duced on the appropriate indicator plates, which contain a substrate for the beta-galactosi- |
|
|||
dase product of this gene that yields a blue-colored product. The second plasmid, initially |
|
|||
Figure 14.13 Use of streptavidin-coated magnetic microbeads for affinity purification of triplex
DNA. Adapted from Ito et al. (1992).
|
|
|
|
|
TRIPLEX-MEDIATED DNA |
CAPTURE |
||
present at much lower concentrations, contained an insert of (AG) |
|
|
|
|
||||
gene in such a way as to disrupt its translation. This results in white colonies after trans- |
|
|||||||
formation |
of |
E. coli. |
Thus the ratio of plasmids in a mixture can be determined just by |
|||||
comparing the number of colonies of each color after transformation (making the reason- |
|
|||||||
able assumption that the transformation efficiencies of the two almost identical plasmids |
|
|||||||
are the same). When a mixture of the two types of DNA was subjected to a single cycle of |
|
|||||||
purification via triplexes formed with magnetic beads containing (TC) |
|
|
|
|
||||
rification |
of the |
(AG) |
n -containing plasmid by a factor |
of 140,000 fold with an overall |
||||
yield |
of |
80%, |
as shown in Table 14.2. Such a purification |
is sufficient |
|
for almost |
any |
|
need. However, if |
necessary the purity could surely be increased much |
further by a sec- |
|
|||||
ond cycle with the magnetic beads. The entire process took only a few hours. Recently |
|
|||||||
Lloyd Smith and coworkers have demonstrated that a similar process can be carried out in |
|
|||||||
only 15 minutes. G- and T-containing sequences can also be used in an analogous way to |
|
|||||||
capture homopurine-homopyrimidine sequences in the presence of Mg |
|
|
|
|
||||
lent cations (Takabatake et al., 1992). |
|
|
|
|
||||
A second test of triplex-mediated capture with magnetic |
microbeads |
is |
shown |
|
||||
schematically in Figure 14.14. Here the challenge was to purify (AG) |
|
|
|
|||||
clones away from the remainder of a chromosome 21-specific library. The original bacte- |
|
|||||||
riophage |
library |
was first subcloned into a plasmid vector. Magnetic purification pro- |
|
|||||
ceeded as described in Figure 14.13. Then the purified DNA was |
used to transform |
|
||||||
coli. |
Individual colonies were picked and screened for the presence of (AG) |
|
|
n |
||||
|
|
|
|
|
|
|
|
|
tween |
each of the |
vector arms and the internal (AG) |
|
n |
sequence. A |
positive signal should |
||
be seen for each |
(AG) |
n -containing clone with one of the two vector |
arms depending on |
|||||
the orientation of the insert (Fig. 14.15). Some of the actual results obtained are shown in |
||||||||
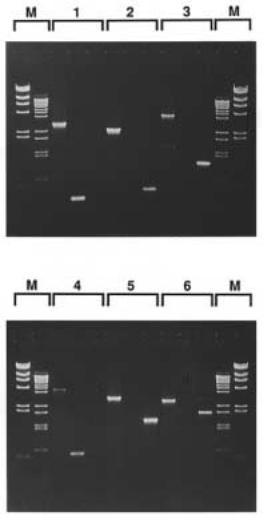
Figure 14.16. The effectiveness of the procedure is quite clear. Overall, 17 of the first 18 |
||||||||
colonies tested showed vector-insert PCR signals, and each of these was different, imply- |
|
|||||||
ing that different genomic (AG) |
n -containing clones had been selected. |
|
||||||
483
E.
by PCR be-
TABLE 14.2 |
Triplex-Mediated Purification of Target |
|
|
||
Plasmids from a Reconstituted Library |
|
|
|||
|
|
|
|
|
|
|
|
Number of Colonies (%) |
|
|
|
|
|
|
|
|
|
|
|
White (pTC45) |
Blue (pUC19) |
||
|
|
|
|
|
|
|
|
Before Enrichment |
|
|
|
|
|
5.0 104 |
1.1 107 |
||
|
|
(0.5) |
|
(99.5) |
|
|
|
After Enrichment |
|
|
|
|
|
4.0 104 |
0.5 |
102 |
|
|
|
(99.9) |
|
(0.1) |
|
|
|
|
|
||
Source: |
Adapted from Ito et al. (1992). |
|
|
||
Note: Plasmids prepared from a reconstituted library were used for |
|
|
|||
transformation |
of |
E. coli |
with or without enrichment by triplex |
||
affinity capture. pTC45 (target) and pUC19 give white and blue |
|||||
colonies, respectively, on indicator plates. In this experiment the |
|
|
|||
enrichment was |
|
1.8-fold10with5 |
a recovery of |
80%. |
|

Figure 14.14 |
Outlined of the procedure used to purify (AG) |
n -containing clones from a chro- |
mosome 21 library.
Figure 14.15 PCR scheme used, after triplex-mediated purification, to screen for clones that contain an (AG) n insert.
TRIPLEX-MEDIATED DNA CAPTURE |
485 |
Figure 14.16 Results of the analysis of a number of triplex-purified clones by the PCR assay described in Figure 14.15. Adapted from Ito et al. (1992).
A third test of triplex capture took advantage of a strain of yeast that Peter Dervan and his co-workers had constructed, containing an insert of a 40-base homopurine-homopy-
rimidine |
sequence |
in |
yeast |
(S. |
cerevisiae) |
chromosome III. This strain was originally |
made to test the efficiency of triplex-mediated DNA cleavage. We were able to obtain and |
||||||
use the same strain to check the efficiency of triplex-mediated DNA capture. Instead of |
||||||
working with yeast genomic DNA, which would suffer considerable shear damage under |
||||||
the conditions used to manipulate the magnetic microbeads, we first subcloned this strain |
||||||
of yeast into a plasmid vector. Then plasmid DNAs were selected using magnetic mi- |
||||||
crobeads containing the appropriate homopyrimidine sequence. In practice, 50% of the |
||||||
clones isolated contained the correct DNA insert as determined by DNA sequencing. The |
||||||
other contaminating clones had a similar, but not identical, sequence that was a natural |
||||||
component |
of the |
yeast |
genome. Presumably |
this |
contaminant could |
have been selected |

486 SEQUENCE-SPECIFIC MANIPULATION OF DNA
against more efficiently by the use of slightly more stringent conditions for triplex formation and washing.
AFFINITY CAPTURE ELECTROPHORESIS
A major limitation with all of the magnetic microbead methods is that the shear damage generated by liquid phase handling of DNA in this way restricts targets to DNAs less than
a few hundred kb in size. To work with larger DNAs, it is necessary that most or all manipulations take place in an anticonvective medium like agarose. This encourages using electrophoresis in agarose to carry DNA past an immobilized triplex capture probe (Ito et
al., 1992). Such an approach has been termed affinity capture electrophoresis (ACE). To test this method, we used streptavidin-containing microbeads (no need for magnets here) embedded in agarose. A DNA sample containing a potential target for triplex formation
with the sequence on the beads was loaded in a sample well and electrophoresed at pH 5
past |
the potential capture |
zone, as shown in Figure 14.17. Then the pH was |
raised to 8, |
|||
and any material captured was released and analyzed in a subsequent electrophoresis step. |
||||||
|
This simple procedure |
works, but at present, its efficiency |
is |
less |
than |
desired. The |
poor |
efficiency arises from |
nonspecific binding between streptavidin |
and |
DNA |
at pH |
5. |
Streptavidin is a protein with an isolectric point of about pH 7. Therefore at pH 5 the protein is positively charged and, as such, binds nonspecifically to DNA. To destabliize these electrostatic interactions, high ionic strength buffers can be used. Such buffers worked very well with the magnetic separations described earlier. However, they lead to serious complications in electrophoretic procedures because the high salt buffers have high conductivity, and as a result there is considerable heating and band broadening. These problems can probably be circumvented by changing the properties of the surface of streptavidin to introduce more negative charges. Since the three-dimensional structure of the protein is known (Chapter 3), the molecular design and engineering needed to accomplish this change in charge should be relatively straightforward.
Figure 14.17 Schematic illustration of a procedure for ACE: Affinity capture electrophoresis. The lightly shaded portion contains gel-embedded immobilized triplex-forming oligonucleotides corre-
sponding to the target of interest.

|
|
|
|
AFFINITY |
CAPTURE ELECTROPHORESIS |
487 |
||
To avoid the problems with the currently available form of triplex-mediated ACE, an |
|
|||||||
alternative capture scheme was developed that could be implemented at neutral pH. This |
|
|
||||||
scheme lacks the attractive generality of triplex |
capture because it is applicable only to |
|
||||||
the ends of DNA fragments, and it requires that information be available about the DNA |
|
|
||||||
sequence at |
one end, or at least |
that a clone |
be available |
that overlaps |
the |
ends |
of the |
|
DNA to be captured. This is a major limitation, in principle, but it is less confining in |
|
|||||||
practice. A major potential application for ACE is to try to purify specific large restriction |
|
|||||||
fragments from complex mixtures. If a linking library that |
corresponds to |
the |
ends |
of |
|
|||
these fragments is available, as described in Chapter 8, then the necessary clones or se- |
|
|||||||
quences will already be in hand. |
|
|
|
|
|
|
|
|
The basic scheme of an alternative ACE procedure, termed end-capture electrophore- |
|
|||||||
sis, is shown in Figure 14.18. In this scheme, the ends of a long DNA duplex are treated |
|
|||||||
with an exonuclease like |
E. coli |
Exonuclease III, or DNA |
polymerase |
in the absence of |
|
|||
triphosphates, to remove a small portion of the 3 |
|
-ends of the duplex and expose the com- |
|
|||||
plementary 5 |
sequence as single strand. The affinity capture medium is made, as before, |
|
||||||
with the sequence complementary to the target. In this case the beads will contain the au- |
|
|||||||
thentic 3 |
-end of the large DNA fragment, synthesized from known sequence or isolated |
|
||||||
as the appropriate half of a linking clone. Capture consists of ordinary duplex formation. |
|
|||||||
The challenge is to find an efficient and nondisruptive way to release the target after it has |
|
|||||||
been captured. An effective way to do this, illustrated in Figure 14.19, is to prepare the |
|
|||||||
capture probe so that it contains the base dU |
instead of T. This still allows efficient, |
|
||||||
sequence-specific strand capture. However, subsequent treatment with Uracil DNA gly- |
|
|
||||||
cosidase, an enzyme that participates in the repair of DNA (Chapter 1), will release the |
|
|||||||
captured target. The same general idea was described in Chapter 4 as |
a |
method |
for |
|
||||
minimizing PCR contamination. |
|
|
|
|
|
|
|
|
Figure 14.20 shows an actual example of end capture electrophoresis. Here, with DNA fragments of the order of 10 kb in size, the method works quite well. It still has not been successful in the capture of much larger targets. This may reflect the much slower rate at which these targets will find a probe during the electrophoresis. Because of excluded vol-
ume effects, the end of a large DNA will be accessible for hybridization only a small fraction of the time. If this is the problem, it should be resolvable in principle by using much slower electrophoresis rates.
Figure 14.18 Schematic procedure for end capture electrophoresis. The appropriate singlestranded probes needed will be easily obtained from half-linking clones (see Chapter 8).

Figure 14.19 An end capture electrophoresis scheme that allows easy release of the captured DNA.
Figure 14.20 Example of the successful implementation of the scheme shown in Figure 14.19. Adapted from Ito et al. (1992). Lane 2 (arrow) shows a band removed by capture. Lane 4 (arrow) shows elution of the captured band.

USE OF BACKBONE ANALOGUES IN SEQUENCE-SPECIFIC DNA MANIPULATION |
|
489 |
||||||
USE OF BACKBONE ANALOGUES IN |
|
|
|
|
|
|
||
SEQUENCE-SPECIFIC |
DNA |
MANIPULATION |
|
|
|
|
|
|
A number of backbone analogues of DNA have been described in recent years. The prin- |
|
|
||||||
ciple force motivating the |
development of such compounds is their presumed usefulness |
|
|
|||||
as DNA or RNA drugs. The |
expectation is that some of these analgoues will be resistant |
|
||||||
to intracellular enzymes that interfere with attempts to use natural RNA or DNA oligonu- |
|
|||||||
cleotides to modulate ordinary cellular processes. We will discuss some of these efforts at |
|
|||||||
the end of the chapter. One feature of a number of the backbone analogues that have been |
|
|||||||
made is replacement of the highly charged phosphodiesters by neutral groups. The sim- |
|
|||||||
plest approach is the use of phosphotriesters, in which the POO |
|
group of the normal |
|
|||||
backbone is replaced by POO-R. The advantage conferred by such a substitution is that, |
|
|||||||
in a duplex or triplex containing such an altered backbone, the natural electrostatic repul- |
|
|||||||
sion between the DNA strands will be minimized. Thus the resulting complexes ought to |
|
|||||||
be much more stable, and one might expect that the backbone analog would preferentially |
|
|
||||||
form duplex or triplex at the expense of the normal strands present in a cell. This is why |
|
|||||||
such compounds are attractive as potential drugs. Normal triplexes with two homopurine |
|
|
||||||
and one homopyrimidine strands, for example, are seriously destabilized at low-salt con- |
|
|||||||
centration because they have three intra-strand sets of electrostatic repulsion compared to |
|
|||||||
only one for the duplex and a separated third strand (Fig. 14.21). |
|
|
|
|||||
Phosphotriesters have a feature that considerably complicates their use. Since each |
|
|||||||
phosphate now has four different substituents, as shown in Figure 14.22, it is optically ac- |
|
|||||||
tive. This means that each alkyl group R can occupy one of two positions on each phos- |
|
|||||||
phate. The resulting number of different stereoisomers for a chain with |
|
n phosphates is 2 |
n . |
|||||
This is a very discouraging prospect for in vivo or in vitro |
studies, and phosphotriesters |
|
||||||
are likely to see rather |
limited use until |
the |
problem of |
synthesizing |
specific |
isomers |
|
|
can be solved. Until this |
has been accomplished, |
attention |
has focused on |
other |
back- |
|
||
bone analogues which |
have the disadvantage of |
being |
less like |
natural nucleic |
acids |
but |
|
|
Figure |
14.21 |
Electrostatic repulsion expected in various DNA structures. |
(a) |
Normal DNA du- |
||
plex. |
(b) |
Normal |
DNA triplex. |
(c) DNA duplex with one strand displaced by |
an uncharged DNA |
|
analog. |
|
(d) DNA |
duplex with one |
strand displaced and a triplex formed at the other strand by |
bind- |
|
ing of two uncharged DNA analogs.
Figure 14.22 Stereoisomeric pair of phosphotriesters.