

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
11 Strategies for Large-Scale DNA Sequencing
WHY |
STRATEGIES ARE |
NEEDED |
|
|
|
|
|
Consider the task of determining |
the sequence of a 40-kb cosmid insert. Suppose |
that |
|||||
each sequence read generates 400 |
base pairs of raw |
DNA sequence. At |
the |
minimum |
|
||
two-fold redundancy needed to assemble a completed sequence, 200 distinct DNA sam- |
|
||||||
ples will have to be prepared, sequenced, and analyzed. At a more typical redundancy of |
|||||||
ten, |
1000 samples |
will have to |
be processed. The |
chain of events |
that |
takes |
the cosmid, |
converts it into these samples, and provides whatever sequencing primers are needed is the upstream part of a DNA sequencing strategy. The collation of the data, their analysis, and their insertion into databases is the downstream part of the strategy. It is obvious that this number of samples and the amount of data involved cannot be handled manually, and
the project cannot be managed |
in an unsystematic fashion. A key part of |
the |
strategy will |
be developing the protocols |
needed to name samples generated along |
the |
way, retain- |
ing an audit of their history, and developing procedures that minimize lost samples, unnecessary duplication of samples or sequence data, and prevent samples from becoming mixed up.
There are several well-established, basic strategies for sequencing DNA targets on the scale of cosmids. It is still not yet totally clear how these can best be adopted for Mb scale sequencing, but it is daunting to realize that this is an increase in scale by a factor of 50, which is not something to be undertaken lightly. The basic strategies range from shotgun, which is a purely random approach, to highly directed walking methods. In this chapter the principles behind these strategies will be illustrated, and some of their relative advantages and disadvantages will be described. In addition we will discuss the relative
merits |
of sequencing genomes and sequencing just the genes, by the use of cDNA li- |
braries. Current approaches for both of these objectives will be described. |
|
SHOTGUN |
DNA SEQUENCING |
This is the method that has been used, up to now, for virtually all large-scale DNA sequencing projects. Typically the genomic (or cosmid) DNA is broken by random shear.
The DNA fragments are trimmed to make sure they have blunt ends, and as such they are subcloned into an M13 sequencing vector. By using shear breakage, one can generate fragments of reasonably uniform size, and all regions of the target should be equally represented. At first, randomly selected clones are used for sequencing. When this becomes
inefficient, because the same clones start |
to |
appear often, |
the set |
of clones already se- |
||||
quenced can |
be pooled |
and hybridized back |
to |
the library |
to |
exclude |
clones that |
come |
from regions |
that have |
already been over sampled (a similar |
procedure |
is described |
later |
|||
361
362 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
in the chapter for cDNA sequencing; see Fig. 11.17). The use of a random set of small
targets ensures that overlapping sequence data will be obtained, |
across the entire target, |
||
and that both strands will |
be sampled. An obvious complication |
that this |
causes is that |
one does not know a priori |
to which strand a given segment of sequence |
corresponds. |
|
This has to be sorted out by software as part of problem of assembling the shotgun clones |
|||
into finished sequence. |
|
|
|
Shotgun sequencing has |
a number of distinct advantages. It |
works, it |
uses only tech- |
nology that is fully developed and tested, and this technology is all fairly easy to automate. Shotgun sequencing is very easy at the start. As redundancy builds, it catches some
of the errors. The number of primers needed is very small. If one-color sequencing or dye terminator sequencing is used, two primers suffice for the entire project if both sides of each clone are examined; only one primer is needed if a single side of each clone is examined. With four-color dye primer sequencing the number of primers needed are eight
and four, respectively.
The disadvantages of shotgun sequencing fall into three categories. The redundancy is very high, definitely higher than more orderly methods, and so, in principle, optimized shotgun sequencing will be less efficient than optimized directed methods. The assembly
of shotgun sequence data is computationally very intensive. Algorithms exist that do the job effectively, such as Phil Green’s phrap program, but these use much computer time. The reason is that one must try, in assembly, to match each new sequence obtained with
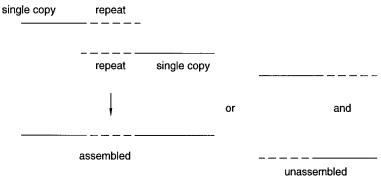
all of the other sequences and their complements in order to compensate for the lack of prior knowledge of where in the target and which strand the sequence derives from. Repeats can confuse the assembly process. A simple example is shown in Figure 11.1. Suppose that two sequence fragments have been identified with a nearly identical repeat.
One possibility is that the two form a contig; they derive from two adjacent clones that overlap at the repeat, and the nonidentity is due to sequencing errors. The other possibility is that the two sequences are not a true contig, and they should not be assembled. The software must keep track of all these ambiguities and try to resolve them, by appropriate statistical tests, as the amount of data accumulates.
Closure is a considerable problem in most shotgun sequencing. Because of the statistics of random picking and the occurrence of uncloneable sequences, even at high redun-
dancy, and even if hybridization is used to help select new clones, there will be gaps. These have to be filled by an alternate strategy; since the gaps are usually small, PCRbased sequencing across them is usually quite effective. A typical shotgun approach to se-
Figure 11.1 Effect of a repeated sequence on sequence assembly.
|
|
DIRECTED SEQUENCING WITH WALKING PRIMERS |
363 |
||||||||||
quencing an entire cosmid will require a total of 400 kb of sequence data (redundancy of |
|
|
|
||||||||||
10) and 1000 four-color sequencing gel lanes. If 32 samples are analyzed at once, and |
|
||||||||||||
three runs can be done per day, this will require 10 working days with a single automated |
|
|
|||||||||||
instrument. In practice, around 20% sample failure can be anticipated, and most laborato- |
|
|
|||||||||||
ries can only manage two runs a day. Therefore a more conservative time estimate is that |
|
|
|||||||||||
a typical laboratory, with one automated instrument at efficient production speed, will re- |
|
|
|||||||||||
quire about 18 working days to sequence a cosmid. This is a dramatic advance from the |
|
|
|||||||||||
rates |
of data acquisition in the very first DNA sequencing |
projects. However, it is |
not yet |
|
|||||||||
a speed at which the entire human genome can be done efficiently. At 8 |
|
|
|
|
104 |
cosmid |
|||||||
equivalents, the human genome would require 4 |
|
|
|
|
|
103 |
laboratory years of effort at this |
|
|||||
rate, even assuming seven-day work weeks. It is not an inconceivable |
task, |
but |
the |
|
|||||||||
prospects will be much more attractive once an additional factor of 10 |
in throughput |
can |
|
|
|||||||||
be achieved. This is basically achievable by the optimization of existing gel-based meth- |
|
||||||||||||
ods described in Chapter 10. |
|
|
|
|
|
|
|
|
|
|
|
||
DIRECTED SEQUENCING WITH WALKING PRIMERS |
|
|
|
|
|
|
|
|
|
|
|
||
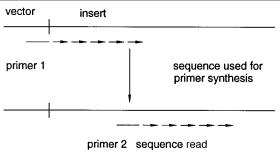
This strategy is the opposite of shotgun sequencing, in that nothing is left to chance. The |
|
||||||||||||
basic idea is illustrated in Figure 11.2. A primer is selected in the vector arm, adjacent to |
|
||||||||||||
the genomic insert. This is used to read as far along the sequence as |
one can in a single |
|
|||||||||||
pass. |
Then the end of the newly determined sequence |
is |
used |
to |
synthesize |
another |
|
||||||
primer, and this |
in turn serves to generate the |
next |
patch of |
|
sequence |
data. |
One |
can |
|
||||
walk in from both ends of the clone, and in practice, this will allow both strands to be |
|
||||||||||||
examined. |
|
|
|
|
|
|
|
|
|
|
|
|
|
The immediate, obvious advantage of primer walking is that only about a twofold re- |
|
|
|||||||||||
dundancy is required. There are also no assembly or closure problems. With good sensi- |
|
|
|||||||||||
tivity in the sequence reading, it is possible to perform walking |
primer sequencing |
di- |
|
|
|||||||||
rectly on bacteriophage or cosmid DNA. This eliminates almost all of |
the steps involved |
|
|
||||||||||
in subcloning and DNA sample preparation. These advantages are |
|
all |
significant, |
and |
|
|
|||||||
would |
make directed priming the method of choice for large-scale |
DNA |
projects if |
a |
|
|
|
||||||
number of the current disadvantages in this approach could be overcome. |
|
|
|
|
|
|
|||||||
The major, obvious, disadvantage in primer walking is that a great deal of DNA syn- |
|
||||||||||||
thesis is required. If 20-base-long primers are used, to guarantee that the |
sequence se- |
|
|||||||||||
lected |
is unique |
in the genome, one must synthesize |
5% of |
the |
total sequence for |
each |
|
||||||
Figure 11.2 Basic design of a walking primer sequencing strategy.
364 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
strand, or 10% of the overall sequence. By accepting a small failure rate, the size of the primer can be reduced. For example, in 45 kb of DNA sequence, one can estimate by Poisson statistics that the probability of a primer sequence not occurring elsewhere, and only at the site of interest is
Primer length |
10 |
11 |
12 |
Unique |
0.918 |
0.979 |
0.998 |
Thus primers less than 12 bases long are not going to be reliable. With 12 mers, one still has to synthesize 6% of the total sequence to examine both strands. This is a great deal of effort, and considerable expense is involved with the current technology for automated oligonucleotide synthesis.
The problem is that existing automated oligonucleotide synthesizers make a minimum of a thousand times the amount of primer actually needed for a sequence determination.
The major cost is in the chemicals used for the synthesis. Since the primers are effectively unique, they can be used only once, and therefore all of the cost involved in their synthesis is essentially wasted. Methods are badly needed to scale down existing automated
DNA synthesis. |
The first multiplex synthesizers built could only make 4 to 10 samples at |
||
a time; this |
is far less than would be needed for large-scale DNA sequencing by primer |
||
walking. Recently |
instruments |
have been described that can make 100 primers at a time |
|
in amounts 10% |
or less of |
what is now done conventionally. However, such instruments |
|
are not yet widely available. In the near |
future primers will be made thousands at a time |
by in situ synthesis in an array format (see |
Chapter 12). |
A second major problem with directed |
sequencing is that each sample must be done |
serially. This is illustrated in Figure 11.3. Until the first patch of sequence is determined, one does not know which primer to synthesize for the next patch of sequence. What this
means is that it is terribly inefficient to sequence only a single sample at once by primer
walking. |
Instead, one must |
work with at least as many samples as there are |
DNA |
se- |
||
quencing |
lanes |
available, |
and ideally one would be processing two to three |
times |
this |
|
amount so |
that |
synthesis |
is |
never a bottleneck. This means that the whole project has to |
||
be orchestrated carefully |
so |
that an efficient flow of samples occurs. Directed |
sequencing |
|||
is not appropriate for a single laboratory interested in only one target. Its attractiveness is for a laboratory that is interested in many different targets simultaneously, or that works on a system that can be divided into many targets that can be handled in parallel.
At present, a sensible compromise is to start a sequencing project with the shotgun strategy. This will generate a fairly large number of contigs. Then at some point one switches to a directed strategy to close all of the gaps in the sequence. It ought to be possible to make this switch at an earlier stage than has been done in the past as walking becomes more efficient and can cover larger regions. In such a hybrid scheme the initial
Figure 11.3 Successive cycles of primer synthesis and sequence determination lead to a potential bottleneck in directed sequencing strategies.

|
PRIMING WITH MIXTURES OF SHORT OLIGONUCLEOTIDES |
365 |
||||||
shotgunning serves to accumulate large numbers of samples, which are then extended by |
|
|||||||
parallel directed sequencing. |
|
|
|
|
|
|
|
|
Three practical considerations enter the |
selection |
of |
particular primer |
sequences. |
|
|||
These can be dealt with by existing software packages that scan the sequence and suggest |
|
|||||||
potentially good primers. The primer must be as specific as possible for the template. |
|
|||||||
Ideally a sequence is chosen where slight mismatches elsewhere on the template are not |
|
|||||||
possible. The primer sequence must occur once and only once in the template. Finally the |
|
|||||||
primer should not be capable of forming stable secondary structure with itself—intramol- |
|
|||||||
ecularly, as a hairpin, or intermolecularly—as a dimer. The former category is particularly |
|
|||||||
troublesome, since, as illustrated in Chapter 10, there are occasional DNA sequences that |
|
|||||||
form unexpectedly stable hairpins, and we do not yet understand these well enough to |
|
|||||||
generalize and be able to identify and reject them a priori. |
|
|
|
|
|
|||
PRIMING WITH MIXTURES OF SHORT OLIGONUCLEOTIDES |
|
|
|
|
|
|||
Several groups have recently |
developed schemes |
to circumvent the difficulties required |
|
|||||
by custom synthesis of large numbers of unique DNA sequencing primers. The relative |
|
|||||||
merits of these different schemes are still not |
fully evaluated, but on the surface, they |
|
||||||
seem quite powerful. The first notion, put forth by William Studier, was to select, arbitrar- |
|
|||||||
ily, a subset of the 4 |
9 possible 9-mers |
and make all of these compounds in advance. The |
|
|||||
chances that a particular 9-mer would be present in a 45-kb cosmid are given below, cal- |
|
|||||||
culated from the Poisson distribution P( |
|
|
|
n ), where |
n |
is the expected number of |
occur- |
|
rences: |
|
|
|
|
|
|
|
|
|
P(O) 0.709 |
P(1) 0.244 |
P( |
1) 0.047 |
|
|||
Therefore, with no prior knowledge about the |
sequence, |
a |
randomly |
selected |
9-mer |
|
||
primer will give a readable sequence about 20% of the time. If it does, as shown in Figure |
|
|||||||
11.4, its complement will also give readable sequence. Thus the original subset of 4 |
9 |
|||||||
|
||||||||
compounds should be constructed of 9-mers and their complements. |
|
|
|
|
||||
One advantage of this approach is that no sequence information about the sample is |
|
|||||||
needed initially. The same set of primers will be used over and over again. As sequence |
|
|||||||
data accumulate, some will contain sites that allow |
priming |
from members |
of the |
pre- |
|
|||
made 9-mer subset. Actual simulations indicate that the whole process could be rather ef- |
|
|||||||
ficient. It is also parallelizable in an unlimited fashion because one can carry as many dif- |
|
|||||||
ferent targets along as one has sequencing gel lanes |
available. An obvious and |
serious |
|
|||||
Figure 11.4 Use of two complementary 9-base primers allows sequence reading in two directions.

366 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
disadvantage of this method is the high fraction of wasted reactions. There are possible ways to improve this, although they have not yet been tried in practice. For example one could pre-screen target-oligonucleotide pairs by hybridization, or by DNA polymerasecatalyzed total incorporation of dpppN’s. This would allow those combinations in which the primer is not present in the target to be eliminated before the time-consuming steps of running and analyzing a sequencing reaction are undertaken.
At about the same time that Studier proposed and began to test his scheme, an alterna-
tive solution to this problem |
was put forth by Wacslaw Szybalski. His original scheme |
was to make all possible 6-mers |
using current automated DNA synthesis techniques. With |
current methods this is quite |
a manageable task. Keeping track of 4096 similar com- |
pounds is feasible with microtitre plate handling robots, although this is at the high end of the current capacity of commercial equipment. To prime DNA polymerase, three adjacent
6-mers are used in tandem, as illustrated in Figure 11.5. They are bound to the target, covalently linked together with DNA ligase, and then DNA polymerase and its substrates
are added. The principle behind this approach is that end stacking (see Chapter 12) will favor adjacent binding of the 6-mers, even though there will be competing sites elsewhere
on the template. Ligation will help ensure a preference for sequence-specific, adjacent 6- mers because ligase requires this for its own function. The beauty of this approach is that the same 6-mer set will be used ad infinitum. Otherwise, the basic scheme is ordinary di-
rected sequencing by primer walking. After each step the new sequence |
must |
be |
scanned |
|
|
to determine which combination of 6-mers should |
be selected as the |
optimal |
choice |
for |
|
the next primer construction. |
|
|
|
|
|
The early attempts to carry out tandem |
6-mer priming seemed |
discouraging. The |
|||
method appeared to be noisy and unreliable. Too |
many of the lanes |
gave unreadable |
se- |
||
quence. This prompted several variants to be developed, as described below. More recently, however, conditions have been found which make the ligation-mediated 6-mer priming very robust and dependable. This approach, or one of the variants below, is worth considering seriously for some of the next generation large-scale DNA sequencing pro-
jects. The key issues that need to be resolved are its generality and the ultimate cost savings it affords against any possible loss in throughput. Smaller-scale, highly multiplexed synthesis of longer primers will surely be developed. These larger primers are always
Figure 11.5 Directed sequencing by ligation-mediated synthesis of a primer, directed by the very template that constitutes the sequencing target.

|
|
|
|
|
PRIMING WITH MIXTURES OF SHORT OLIGONUCLEOTIDES |
367 |
||
likely to perform slightly better than ligated mixtures of smaller compounds because the |
|
|||||||
possibility of using the 6-mers in unintended orders, or using only two of the three, is |
|
|||||||
avoided. There is enough experience with longer primers to know how to design them to |
|
|||||||
minimize chances of failure. Similar experience must be gained with 6-mers before an ac- |
|
|||||||
curate evaluation of their relative effectiveness can be made. |
|
|||||||
Two more recent attempts to use 6-base oligonucleotide mixtures as primers avoid the |
|
|||||||
ligation step. This saves time, and it also saves any potential complexities arising from |
|
|||||||
ligase side reactions. Studier’s approach is to add the 6-mers to DNA in the presence of a |
|
|||||||
nearly |
saturating |
amount |
of |
single-stranded DNA binding protein (ssb). This protein |
|
|||
binds to DNA single strands selectively and cooperatively. Its effect on the binding of sets |
|
|||||||
of small complementary short |
oligonucleotides is shown schematically in Figure 11.6. |
|
||||||
The key point is the high cooperativity |
of ssb to single-stranded DNA. What this does is |
|
||||||
to make configurations in which 6-mers are bound at separate sites less favorable, ener- |
|
|||||||
getically, than configurations in which they are stacked together. In essence, ssb coopera- |
|
|||||||
tivity is translated into 6-mer DNA binding cooperativity. This cuts down on false starts |
|
|||||||
from individual or |
mispaired |
6-mers. An unresolved issue is how much mispairing still |
|
|||||
can go on under the conditions used. We have faced before the problem in evaluating this; |
|
|||||||
there are many possible 6-mers, |
and many |
more possible sets of 6-mer mixtures, so only |
|
|||||
a minute fraction of these possibilities has yet been tried experimentally. One simplifica- |
|
|||||||
tion is the use of primers with 5-mC and 2-aminoA, instead of C and A, respectively. This |
|
|||||||
allows five base primers to be substituted for six base primers, reducing the complexity of |
|
|||||||
the overall set of compounds needed to only 1024. |
|
|||||||
An |
alternative approach |
to |
the problem has been developed by Levy Ulanovsky. He |
|
||||
has shown that conditions can |
be |
found where it is possible to eliminate both the ligase |
|
|||||
and the ssb and still see highly |
reliable priming. Depending on the particular sequence, |
|
||||||
some five base primers can be substituted for six base primers. The effectiveness of the |
|
|||||||
approach appears to derive from the intrinsic preference of DNA polymerase for long |
|
|||||||
primers, so a stacked set is preferentially utilized even though individual members of the |
|
|||||||
set may bind to the DNA template |
elsewhere alone or in pairs. It remains to be seen, as |
|
||||||
each of |
these approaches |
is developed further, which will prove to be the most efficient |
|
|||||
and accurate. Note that in any directed priming scheme, for a 40-kb cosmid, one must se- |
|
|||||||
quence 80 kb of DNA. If a net of 400 new bases of high-quality DNA sequence can be |
|
|||||||
completed per run, it will take 200 steps |
to |
complete the walk. As a worst-case analysis, |
|
|||||
one may have to search the 3 |
|
|
|
-terminal 100 bases of a new sequence to find a target suit- |
|
|||
able for priming. Around |
500 |
base pairs |
of |
sequence must actually be accumulated per |
|
|||
run to achieve this efficiency. Thus a directed priming cosmid sequencing project is still a considerable amount of work.
Figure 11.6 Use of single-stranded binding protein (ssb) to improve the effectiveness of tandem, short sequencing primers.
368 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
ORDERED SUBDIVISION OF DNA TARGETS
There are a number of strategies for DNA sequencing that lie between the extreme randomness of the shotgun approach and the totally planned order of primer walking. These
schemes |
all have in common a set of cloning or physical purification steps that divides |
the DNA |
target into samples that can be sequenced and that also provides information |
about their location. In this way a large number of samples can be prepared for sequenc- |
|
ing in parallel, which circumvents some of the potential log jams that are undesirable fea- |
|
tures of |
primer walking. At the same time from the known locations one can pick clones |
to sequence in an intelligent, directed way and thus cut down substantially on the high re- |
|
dundancy of shotgun sequencing. One must keep in mind, however, that the cloning or separation steps needed for ordered subdivision of the target are themselves quite labor intensive and not yet easily automated. Thus there is no current consensus on which is the
best approach. One strategy used recently |
to |
sequence |
a number |
of |
small |
bacterial |
|
genomes is the creation of a dense (highly redundant) set of clones in a sequence-ready |
|||||||
vector—that is, a vector like a cosmid which allows direct sequencing without additional |
|||||||
subcloning. The sequence at both ends of each of the |
clones is |
determined |
by |
using |
|||
primers from known vector sequence. As more |
primary DNA |
sequence |
data |
become |
|
||
known and available in databases, the chances that one or both ends |
of each clone |
will |
|||||
match previously determined sequence increase considerably. Such database matches, as |
|
||||||
well as any matches among the set of clones |
themselves, |
accomplish |
two |
purposes |
at |
||
once. They order the clones, thus obviating much or all of the need for prior mapping ef- |
|||||||
forts. In addition they serve as staging points |
for the construction |
of |
additional |
primers |
|||
for directed sequencing. Extensions of the use of sequence-ready clones |
are discussed |
||||||
later in the chapter. |
|
|
|
|
|
|
|
Today, there is no proven efficient strategy to subdivide a mega-YAC for DNA se- |
|||||||
quencing. Some strategies that have proved effective for subdividing smaller clones are |
|||||||
described in the next few sections. A megabase YAC is, at an absolute minimum, going to |
|||||||
correspond to 25 cosmids. However, there is |
no reason to think that this is |
the |
optimal |
||||
way to subdivide it. Even if direct genomic sequencing from YACs proves feasible, one |
|||||||
will want to have a way of knowing, at least approximately, where in the YAC a given |
|||||||
primer or clone is located, to avoid becoming hopelessly confused by interspersed repeat- |
|||||||
ing sequences. However, for large YACs there is an additional problem. As described |
|||||||
elsewhere, these clones are plagued by rearrangements and unwanted insertions of yeast |
|||||||
genomic DNA. For sequencing, the former problem may be tolerable, but |
not |
the latter. |
|||||
To use mega-YACs for direct sequencing, we would first need to develop methods to re- |
|||||||
move or ignore contaminating yeast genomic DNA. |
|
|
|
|
|
|
|
TRANSPOSON-MEDIATED DNA SEQUENCING
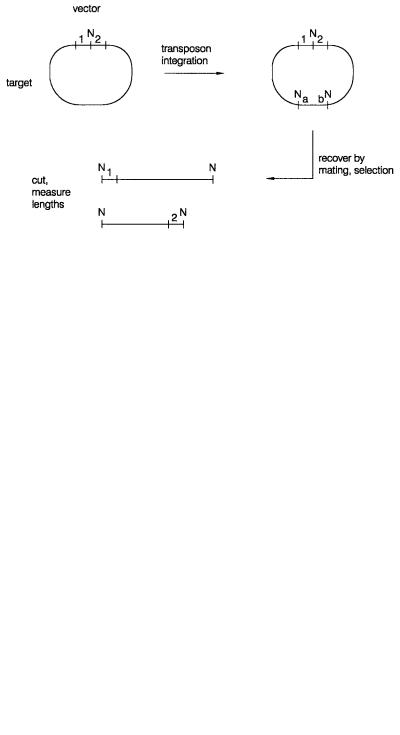
The structure of a typical transposon is shown in Figure 11.7. It has terminal repeated se-
quences, a selectable marker, and codes for a transposase that allows |
the transposon to |
hop from one DNA molecule (or location) to another. The ideal transposon for DNA se- |
|
quencing will have recognition sites for a rare restriction enzyme, |
like Not I, located at |
both ends, and it will have different, known, unique DNA sequences close to each end. A |
|
scheme for using transposons to assist the DNA sequencing of clones in |
|
in Figure 11.8. The clone to be sequenced is transformed into |
a transposon-carrying |
E. coli |
is shown |
E.
TRANSPOSON-MEDIATED DNA SEQUENCING |
369 |
Figure 11.7 |
Structure of a typical transposon useful for DNA sequencing. |
N is a Not I site; |
a and |
b are unique transposon sequences. Arrow heads show the terminal inverted repeats of the transpo- |
|
|
|
son. |
|
|
|
coli. |
It is present in an |
original vector containing a single infrequent |
cutting site |
(which |
|||
can be the same as the ones |
on the transposon) and two unique known DNA sequences |
|
|||||
near to the vector-insert boundary. The transposon may jump into the cloned DNA. If it |
|
||||||
does so, this appears to occur approximately at random. The nonselectivity of transposon |
|
||||||
hopping is a key assumption |
in the potential effectiveness of this strategy. From results |
||||||
that have been obtained thus far, the randomness of transposon insertions seems quite suf- |
|
||||||
ficient to make this approach viable. |
|
|
|
||||
|
DNA from cells in which transposon jumping has potentially occurred can be trans- |
|
|||||
ferred to a new host cell that does not have a resident transposon, and recovered by selec- |
|||||||
tion. The transfer can be done by mating or by transformation. Since the transposon car- |
|
||||||
ries a selectable marker, and the vector |
which originally contained the |
clone carries |
a |
||||
second selectable marker, there will be no difficulty in capturing the desired transposon |
|
||||||
insertions. The goal now is to find a large set of different insertions and map them within |
|||||||
the |
original cloned |
insert |
as efficiently as possible. The infrequent cutting sites placed |
||||
into |
the |
transposon |
and the |
vector greatly |
facilitate this analysis. DNA is |
prepared from |
|
all of the putative transposon insertions, cut with the infrequent cutter, and analyzed by ordinary agarose gel electrophoresis, or PFG, depending on the size of the clones. Each insert should give two fragments. Their sizes give the position of the insert. There is an ambiguity about which fragment is on the left and which is on the right. This can be resolved if the separated DNAs are blotted and hybridized with one of the two unique se-
quences originally placed in the vector (labeled 1 and 2 in Fig. 11.8).
Figure 11.8 |
One strategy for DNA sequencing by transposon hopping. |
a, b, and N are as defined |
in Figure 11.7; 1 and 2 are vector unique sequences.
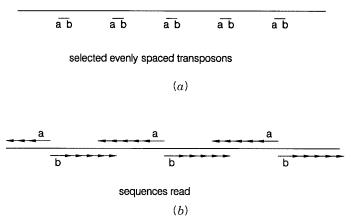
370 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
Figure 11.9 |
DNA sequencing from a |
set of transposon insertions. |
(a ) Chosing a set of properly |
spaced transposons. ( |
b ) Use of |
transposon sequences as primers. |
|
Now the position of the transposon insertion is known. One can select samples that ex- |
|
||||||
tend through the target at intervals than |
can be spanned by sequencing, |
as |
shown |
in |
|
||
Figure 11.9. To actually determine the sequence, one can use the two unique sequences |
|
||||||
within |
the transposon as primers. These will always allow sequence to be |
obtained |
in |
|
|||
both directions extending out from the site of the transposon insertion (Fig. 11.9). One |
|
||||||
does not a priori know the orientation of the two sequences primed from the transposon |
|
|
|||||
relative to the initial clone. However, this |
can be determined, if necessary, by |
reprobing |
|
||||
the infrequent-cutter digest fingerprint of the clone with one of the transposon unique se- |
|
||||||
quences. In practice, though, if a sufficiently dense set of transposon inserts is selected, |
|
||||||
there will be sufficient sequence overlap to assemble the sequence and confirm the origi- |
|
||||||
nal map of the transposon insertion sites. |
|
|
|
|
|
||
DELTA |
RESTRICTION |
CLONING |
|
|
|
|
|
This |
is a relatively straightforward procedure that allows DNA sequences |
from |
vector |
|
|||
arms to be used multiple times as sequencing primers. It is a particularly efficient strategy |
|
||||||
for sampling the DNA sequence of a target in numerous places and for subsequent, paral- |
|
|
|||||
lel, directed primer sequencing. Delta restriction cloning requires that the target of inter- |
|
||||||
est be contained near a multicloning site in the vector, as shown in Figure 11.10. The var- |
|
||||||
ious enzyme recognition sequences in that site are used, singly, to digest the clone, and all |
|
||||||
of the resulting digests are analyzed by gel |
electrophoresis. One looks for |
enzymes |
that |
|
|||
cut the clone into two or more fragments. These enzymes will have cutting |
sites within |
|
|||||
the insert. From the size of the fragments |
generated, one can determine approximately |
|
|||||
where in the insert the internal cleavage site is located. Those cuts that occur near regions |
|
||||||
of the insert where sequence is desired can be converted to targets for sequencing by di- |
|
||||||
luting the cut clones, religating them, and transforming them back into |
|
|
E. coli. |
The effect |
|||
of the cleavage of religation is to drop out from the clone all of the DNA between the |
|
||||||
multicloning site |
and the internal restriction |
site. This brings the vector arm containing |
|
||||