

|
|
|
DEALING |
WITH UNEVEN |
cDNA DISTRIBUTION |
381 |
ase. This procedure is generally preferred over the S1 nuclease method because it tends to |
|
|||||
produce longer, more intact cDNAs. |
|
|
|
|
|
|
An unfortunate fact about many cDNA clones is that they are biased toward the 3 |
|
-end |
||||
of the original message because of the poly A capture and the oligo-T priming used to |
|
|||||
prepare them. The true 5 |
-end is often missing and needs to be found in other clones or in |
|
||||
the genome. Some attempts have been made to take advantage of the specialized cap |
|
|||||
structure at the 5 |
-end of eukaryotic mRNAs to purify intact molecules. One possibility is |
|
||||
to try to produce high-affinity monoclonal antibodies specific for this cap structure. More |
|
|||||
effective has been the use of an enzyme called tobacco pyrophosphatase |
|
. This cleaves off |
||||
the cap to leave an ordinary 5 |
-phosphate-terminated DNA strand that then can serve as a |
|
||||
substrate in a ligation reaction, which can be used to add a known sequence. This known |
|
|||||
sequence will serve as the staging site for subsequent PCR amplification. Several differ- |
|
|||||
ent Japanese groups have recently perfected such strategies to the point where |
5 |
-end- |
||||
containing cDNA libraries can now be made quite reliably. |
|
|
|
|
||
DEALING WITH UNEVEN cDNA DISTRIBUTION |
|
|
|
|
||
With relatively rare exceptions like rDNAs, genes in the genome are in approximately a |
|
|||||
1:1 ratio. In contrast, the relative amount of mRNAs present in a typical cell extends over |
|
|||||
a range of more than 10 |
|
5. This leads to very serious biases in most cDNA libraries. These |
|
|||
will tend to be overrepresented |
with a relatively small numbers of different |
high-fre- |
|
|||
quency clones. In addition existing cloning methods will tend to bias the libraries toward |
|
|||||
short mRNAs. If one attempts to sequence cDNAs at random from a library, in most cases |
|
|
||||
the high copy number clones will be re-sequenced over and over again, while most rare |
|
|||||
mRNAs will never be sampled. It is important to stress that the problems of random se- |
|
|||||
lection and library biases seriously interfere with |
genomic DNA |
sequencing projects, |
|
|||
even though one is starting with an almost uniform sample of the genome. With cDNAs |
|
|||||
these problems are much more serious and must be dealt with directly and forcefully. |
|
|
||||
One simple approach to systematic sequencing of cDNA libraries is shown in Figure |
|
|||||
11.17. One starts with an arrayed library. A small number of clones, say 100, are selected |
|
|||||
and sequenced. All of the |
sequenced |
clones are pooled, |
labeled, and hybridized back to |
|
||
Figure 11.17 |
Basic scheme for sequencing an arrayed cDNA library, and periodically screening |
|
the |
library to detect repeats of clones that have already been sequenced. The schematic array shown |
|
has |
only 56 clones; a real array would have tens to hundreds of times more. |
|
382 |
STRATEGIES FOR LARGE-SCALE DNA SEQUENCING |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
the library. Any clones that are detected by this hybridization are not included in the next |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
set of 100 to be sequenced. By continuing in this way, most duplication can be avoided. |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
Unfortunately, there will also be a tendency |
|
to |
discard |
cDNAs from |
gene |
|
families, |
so |
|
|
|
|
|
|
|
|
|
||||||||||
many of the members of these families will be underrepresented or missed. As an alterna- |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
tive to handling the clones as arrays, one can carry out |
a solution |
hybridization |
of the |
|
|
|
|
|
|
|
|
||||||||||||||||
entire cDNA library with an excess of sequenced clones, discard all the samples that |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
hybridize, and regrow the remainder. This effectively replaces a screen by a physical |
|
|
|
|
|
|
|
|
|||||||||||||||||||
selection process. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
A more complex approach to compensating for uneven cDNA distribution is to try to |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
equalize or normalize the library. The distribution of mRNAs in a typical cell is shown in |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
Table 11.1. Roughly speaking there are three classes of messages: a few very common |
|
|
|
|
|
|
|||||||||||||||||||||
species, then approximately equal total amounts of species |
20 |
times |
more |
rare, |
and |
|
|
|
|
|
|
|
|
|
|||||||||||||
species another factor of 20 rarer still. The goal is to try to even out these differences. The |
|
|
|
|
|
|
|||||||||||||||||||||
approach used is to anneal the library to itself and remove all the double-stranded species |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
that are formed. We will do this by allowing the reannealing to occur at a very high |
|
|
|
|
|
|
|
|
C |
0 t: |
|||||||||||||||||
Typically |
C |
0 t 250 is |
used. From |
|
Chapter 3, |
we |
can |
write |
for |
the fraction |
of |
|
single |
|
|
|
|
|
|||||||||
strand remaining in a hybridization: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
f |
|
1 |
|
|
|
|
|
1 |
|
|
|
|
|
|
C 0 t1/2 |
|
|
|
|
|
||||
|
|
|
1 n |
C |
|
t k /2N |
1 C |
|
t/C |
|
|
C |
|
t1/2 C |
|
t |
|
|
|||||||||
|
|
|
s |
0 |
|
0 |
0 |
t1/2 |
0 |
0 |
|
|
|||||||||||||||
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
where all the quantities in this equation have |
been defined in Chapter |
3. When |
|
|
|
|
|
|
|
|
C 0 t |
||||||||||||||||
C 0 t |
1/2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, we can approximate this result as |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
f |
C 0 t1/2 |
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
s |
|
C |
0 |
t |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Note |
that |
the |
C 0 t1/2value for a |
given |
sequence |
depends |
on |
the |
ratio of genome |
size, |
|
|
|
|
N, |
||||||||||||
and the number of times the sequence is represented, |
|
|
|
|
|
|
|
|
|
n. |
For a cDNA library, |
|
|
N |
is the total |
|
|||||||||||
complexity of the DNA sequences represented in the library, and |
|
|
|
|
|
|
|
|
|
|
|
|
n is the number of times |
|
|||||||||||||
a given sequence is represented. Thus, for highly frequent cDNAs, |
|
|
|
|
|
|
|
|
|
|
|
N |
/n will be |
small so |
|
||||||||||||
that |
the |
C 0 t1/2will be small, and these species will |
renature relatively more rapidly. Note |
|
|
|
|
|
|||||||||||||||||||
that the amount of a particular cDNA remaining after extensive annealing will be propor- |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
tional to its original abundance |
|
|
|
n |
and to |
its |
hybridization |
rate, |
which |
will |
scale as |
|
|
|
N |
/n. |
|||||||||||
Thus, at very long times in the reaction, a relatively even distribution of cDNAs should be |
|
|
|
|
|
|
|
|
|||||||||||||||||||
produced. We can evaluate the expected results for an attempt |
to |
normalize |
the typical |
|
|
|
|
|
|
|
|
|
|||||||||||||||
cell mRNAs shown in Table 11.1. This is given in Table 11.2. |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
TABLE 11.1 |
Distribution of mRNA in a Typical Cell |
|
|
|
|
|
|
Species |
Percent |
Number of |
Relative |
C |
0 |
t1/2 |
|
|
|
Species |
Frequency |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
Common |
10 |
10 |
330 |
0.08 |
|
|
|
Medium |
45 |
1000 |
15 |
1.7 |
|
|
|
Rare |
45 |
15,000 |
1 |
25. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DEALING WITH UNEVEN cDNA |
DISTRIBUTION |
383 |
TABLE 11.2 |
Effect of Self-Annealing a cDNA Library |
|
|
|
|||
|
|
|
|
|
|
||
Class |
f at C |
0 |
t 250 |
Initial Frequency |
Equalized Frequency |
||
|
s |
|
|
|
|
|
|
Common |
3 |
10 4 |
330 |
9.9 |
10 2 |
||
Medium |
7 |
10 3 |
15 |
1.0 |
10 1 |
||
Rare |
9 |
10 2 |
1 |
9.0 |
10 2 |
||
The predictions in Table 11.2 look very encouraging. However, a serious potential problem is that one has to discard most of the library in order to achieve this result. PCR or efficient recloning must be used to recover the cDNA clones which have not self-annealed.
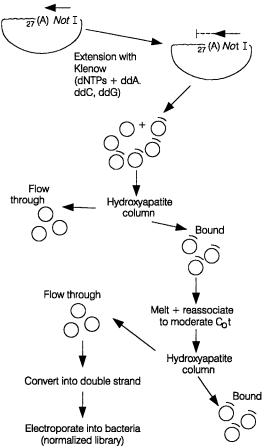
An actual scheme for efficient cDNA normalization is shown in Figure 11.18. This has been developed by Bento Soares, Argiris Efstratiadis, and their collaborators. It is designed to avoid the preferential loss of long cDNA clones during the self-annealing, and
also to avoid the loss of cDNAs from closely related gene families. Long clones would be
Figure 11.18 A relatively elaborate scheme for cDNA normalization that attempts to prevent a bias against shorter cDNAs and the loss of cDNAs from gene families. Adapted from Soares et al.
(1994).
384 |
|
STRATEGIES FOR LARGE-SCALE DNA SEQUENCING |
|
|||
|
TABLE |
11.3 |
cDNA Library Normalization |
|
|
|
|
|
|
|
|
|
|
|
Probes |
|
Original Library |
Normalized (HAP-FT) |
HAP-Bound |
|
|
|
|
|
|
|
|
|
Human C |
0 t 1 DNA |
10% |
2% |
2.6% |
|
|
Elongation factor 1a |
4.6% |
0.04% |
3.7% |
||
|
a-Tubulin |
|
3.7%–4.4% |
0.045% |
6% |
|
|
b-Tubulin |
|
2.9% |
0.4% |
0.85% |
|
|
Mitochondrial 16S rRNA |
1.3% |
1% |
|
||
|
Myelin basic protein |
1% |
0.09% |
|
||
|
g-Actin |
|
0.35% |
0.1% |
1.3% |
|
|
Hsp 89 |
|
0.4% |
0.05% |
0.14% |
|
|
CH13-cDNA#8 |
0.009% |
0.035% |
|
||
|
|
|
|
|
||
|
Source: |
Data adapted from Soares et al. (1994). |
|
|
||
lost if entire cDNA sequences were used |
for |
hybridization |
because, |
as |
described |
in |
||||||
Chapter 3, their rate constants for duplex formation ( |
|
|
|
|
|
|
k ’s) are larger and because there are |
|||||
|
|
|
|
|
|
|
|
|
|
|
2 |
|
more places to nucleate potential duplex. The trick used in the scheme of Figure 11.18 is |
||||||||||||
to start with single-stranded cDNA clones and produce |
a short |
duplex |
at |
the 3 |
|
-end of |
||||||
each cDNA clone by primer extension in the presence |
of |
chain terminators. By |
focusing |
|
||||||||
on this region, one |
will ensure that the new |
strands |
synthesized |
preferentially come from |
||||||||
3 noncoding flanking regions where even closely related genes have significant diver- |
||||||||||||
gence, since the sequences are not translated and presumably |
have little |
function. Any |
||||||||||
cDNAs that have not successfully templated the synthesis of a short duplex are discarded |
|
|||||||||||
by chromatography on hydroxyapatite, which specifically binds only duplexes, under the |
|
|||||||||||
conditions used. These duplex-containing clones are then eluted, melted, and allowed to |
||||||||||||
self-anneal to high |
C 0 t. Now any |
clones |
with duplexes are |
removed, |
and the |
clones that |
||||||
have remained as single strands represent the normalized library. These are then amplified |
||||||||||||
and sequenced. |
|
|
|
|
|
|
|
|
|
|
|
|
Some actual results using the scheme of Figure 11.18 are given in Table 11.3. It is ap- |
||||||||||||
parent that the equalization is far from perfect. However, it represents a major improve- |
||||||||||||
ment over nonnormalized libraries, and materials made in this |
way |
are |
currently being |
|||||||||
used extensively for cDNA sequencing. Two additional schemes for cDNA normalization |
|
|||||||||||
are described in Box 11.1. It is not clear at the present time just which schemes will ulti- |
||||||||||||
mately be widely adopted. |
|
|
|
|
|
|
|
|
|
|
|
|
LARGE-SCALE cDNA SEQUENCING |
|
|
|
|
|
|
|
|
|
|
|
|
In the past three years at least five separate efforts have |
been made to collect massive |
|||||||||||
amounts of cDNA sequence. One of these is a collaboration between the Institute for |
||||||||||||
Genome Research (TIGR) and Human Genome Sciences, Inc. At least initially, this effort |
||||||||||||
took an anatomical approach. Libraries of cDNAs from as many different major tissues as |
|
|||||||||||
possible were collected, and large numbers of clones from each of these were sequenced. |
|
|||||||||||
The second approach was orchestrated by Incyte Pharmaceuticals, Inc. Here the emphasis |
||||||||||||
was on cell physiology. Sets of cDNA libraries were collected |
from |
pairs |
of |
cells |
in |
|||||||
known, related physiological states, such |
as |
activated or |
unactivated |
macrophages. |
A |
|||||||
fixed number of cDNAs, 5000 in the earliest studies, was randomly selected |
for |
each of |
||||||||||
the cell pairs and sequenced. In this way information was obtained |
about the frequencies |
|
||||||||||
of common cDNAs in |
addition to the sequence |
information from all |
classes |
of |
cDNAs. |
|
||||||

LARGE-SCALE cDNA SEQUENCING |
385 |
BOX 11.1
ALTERNATE SCHEMES FOR NORMALIZATION OF cDNA LIBRARIES
Two different schemes for the production of normalized cDNA libraries have been described. The first, proposed by Sherman Weissman and coworkers, is shown schemati-
cally in Figure 11.19. First PCR is used to amplify cloned cDNAs. Then, as in the Soares and Efstratiadis scheme described in the text, hydroxylapatite fractionation is
used to deplete a reaction mixture of double-stranded products. Next a nested set of PCR primers is used to amplify the single-stranded material that survives hydroxyapatite. Finally this material is cloned to make the normalized library. A survey of typical results is given in Table 11.4.
The scheme developed by Michio Oishi is quite different (Fig. 11.20). Here cDNA immobilized on microbeads is annealed to a vast excess of mRNA from the same
source. Under these conditions the kinetics of hybridization become pseudo–first order as described in Chapter 3. The highly overrepresented components in the mRNA will actually deplete the corresponding cDNAs below the level of normalization. The resulting cDNA library will be enriched for rare cDNA sequences. A survey of typical results is given in Table 11.5.
Figure 11.19 |
A relatively simple scheme for cDNA normalization. Adapted from |
Sankhavaram et al. (1991).
(continued)

386 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
BOX 11.1 |
(Continued) |
|
|
|
TABLE 11.4 |
Effect of Normalization on a cDNA Library |
|
|
|
|
|
|
|
|
|
|
|
Number of Clones Identified per 100,000 Plaques |
|
|
|
|
|
|
Probe |
STH |
NSTH I |
NSTH II |
|
|
|
|
|
|
R-DNA |
30,000 |
|
94 |
12 |
Blur-8 |
800 |
|
450 |
360 |
-actin |
110 |
37 |
NT |
|
HLA-H |
104 |
|
80 |
10 |
CD4 |
28 |
|
37 |
12 |
CD8 |
15 |
|
55 |
12 |
Oct-1 |
9 |
NT |
8 |
|
-globin |
7 |
|
NT |
10 |
c-myc |
5 |
|
NT |
11 |
TCR |
5 |
|
NT |
8 |
TNF- |
5 |
NT |
6 |
|
-fodrin |
3 |
|
NT |
9 |
|
|
|
|
|
Source: |
From Patanjali et al. (1991). |
|
|
|
Note: cDNAs present at various levels of abundance in STH library become almost identically abundant in |
|
|||
the normalized (NSTH) libraries. Increased reassociation times, as indicated by the increased |
C 0 t value, ren- |
|||
der better normalized libraries. NT, not tested. For NSTH I the |
C 0 t value was 41.7 mol-s /liter, and for NSTH |
|||
II the C 0 t value was 59.0 mol-s /liter. |
|
|
||
Figure 11.20 A scheme for the preferential enrichment of rare cDNAs. Adapted from Sasaki et al. (1994a).
(continued)

BO |
X 11.1 |
(Continued) |
T ABLE |
11.5 |
Change of the Pr |
oportion of cDN |
A Clones Bef |
or e and After Self-Hybridization |
|
|
|
|
Probe |
|
|
Input |
a |
Before |
Percentage |
After |
Percentage |
After /Before |
|
|
|
|||||||
Rabbit -globin |
1 |
111/10,500 |
1.067 |
5/55,000 |
|
X174 Hae |
III 0.6 kb |
0.01 |
2 /30,000 |
0.0067 |
8/35,000 |
X174 Hae |
III 0.9 kb |
0.01 |
1/30,000 |
0.0033 |
6/30,000 |
neo r |
|
0.0001 |
0/250,000 |
0.0004 |
2 /250,000 |
-actin |
|
|
54 /10,000 |
0.54 |
2 /10,000 |
IL-4 |
|
|
0/320,000 |
0.0003 |
3/320,000 |
IL-2 |
|
|
0/320,000 |
0.0003 |
0/320,000 |
b
b
0.009 |
0.086 |
0.023 |
3.43 |
0.02 |
6 |
0.0008 |
2 |
0.02 |
0.037 |
0.0009 |
3 |
0.0003 |
— |
Sour ce: |
Adapted from Sasaki et al. (1994). |
|
a Percent of total RN |
A (w/ w). |
|
b The positi |
ve clones were confirmed by sequencing approximately 300 bp of the inserts. |
|
387

388 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
The frequency information, when pairs of cells are compared, is often quite interesting,
and it suggests potential functional roles for |
a number of newly discovered genes in the |
|||
libraries. Incyte has |
termed |
such |
comparisons transcript imaging. A third large-scale |
|
cDNA sequencing effort |
is the |
Image |
consortium |
involving several academic or govern- |
ment laboratories and Merck, Inc. Here normalized libraries are serving as the source of
clones for sequencing, and the goal is to collect |
at |
least one |
representative cDNA se- |
quence from all human genes. |
|
|
|
At present, the sequencing of human cDNAs is being carried out in large laboratory ef- |
|||
forts like the three just described as well as many |
smaller, more focused efforts. Within |
||
each laboratory the amount of duplication seen thus |
far |
has been |
relatively small. Thus |
the early course of the cDNA sequencing strategy appears to be very effective. At what point it will peter out into a morass of duplicate clones is unknown. It is also really unclear what fraction of the total amount of genes will actually be found first through their cDNAs. The tissues used for the majority of these studies are those where large numbers
of different genes are expected to be active. These include early embryos, hytidaform moles, which are differentiated but disordered tumors with many different tissue types, liver, and a number of parts of the brain. Whether many specialized tissues will have to be looked at to get genes expressed only in these tissues, or whether there is a broad enough low-level synthesis of almost any mRNA in one or more of the common tissues to let all genes be found there, is an issue that has not yet been answered. One way to try to extend the cDNA approach to find all of the human genes is described in Box 11.2
BOX 11.2
PREPARATION AND USE OF hncDNAs
A major purpose of making ordered libraries is to assist the finding and mapping of genes. Eugene Sverdlov and his colleagues in Moscow have developed an efficient procedure for preparing chromosome-specific hncDNA libraries. Their method is an elaboration of the scheme originally described by Corbo et al. (1990). The procedureis
outlined in Figure 11.21. It uses an initial Alu-primed |
PCR |
reaction to make an |
|
hncDNA copy of the hnRNA produced in a hybrid cell containing |
just |
the chromo- |
|
some of interest. (See Chapter 14 for details about the Alu |
repeat and Alu-specific |
||
PCR primers.) The resulting DNA is equipped with an oligo-G |
tail, and then a first |
||
round PCR is carried out using an oligo-C containing primer and an Alu primer. Then |
|||
a second round of PCR is done with a nested Alu primer. The PCR primers are also |
|||
designed so that the first round introduces one restriction site and the second round an- |
|||
other. The resulting products are then directionally cloned into a vector requiring both |
|||
restriction enzyme cleavage sites. In studies to date, Sverdlov and his coworkers have |
|||
found that this scheme produces a diverse set of highly enriched human cDNAs. Since |
|||
these come from Alu’s in hnRNA, they will contain introns, but they can be used to lo- |
|||
cate genes on the chromosome equally well if not better than conventional cDNAs. |
|
||
Note that the hncDNA clones as produced by the Sverdlov method actually contain |
|||
substantial amounts of intronic regions. This means that they |
will |
be more |
effective |
than the ordinary cDNAs in dealing with gene families and in avoiding cross-hy- bridization with conserved exonic sequences in rodent-human cell hybrids.
(continued)

WHAT IS MEANT BY A COMPLETE GENOME SEQUENCE? |
389 |
BOX 11.2 |
(Continued) |
Figure 11.21 |
Steps involved in making an hncDNA library. Interspersed repeat elements in |
|
|
|
||
hnRNA are represented in the upper line by solid boxes |
(R ). Arrows |
indicate |
primers. Vertical |
|||
lines crossing the arrows symbolise the primers with sites for |
Eco |
R I |
and |
Bam |
H I restriction |
|
endonucleases ( |
E ) or ( B ). EC is 5 GAGAATTC(C)203 |
. The open boxes with |
similar |
symbols |
||
represent sequences corresponding to primers that are included in PCR products P-1 and P-2. Provided by Eugene Sverdlov.
WHAT IS MEANT BY A COMPLETE GENOME SEQUENCE?
If the strictest definition is used for a complete genome sequence, namely every base on every chromosome in a cell has been identified, then it is probably safe to say that we will never accomplish this. This is not to say that the task couldn’t be accomplished in principle; it could be, but for several reasons it is a foolish task, at least for the human genome. The human genome is quite variable. This will be discussed in more detail later. Suffice it to say here that there are millions of differences in DNA sequence between the set of two homologous chromosomes in a diploid cell. Unless one could separate these into sepa-
rate, cloned libraries, inevitable confusion will develop as to which homolog one is on. Hybrid cells make these separations for us, and libraries made from chromosomes of hy-
390 STRATEGIES FOR LARGE-SCALE DNA SEQUENCING
brid cells are major candidates for eventual large-scale sequencing. However, because of the history behind the construction of such hybrids, we rarely have separate clones of two homologous chromosomes from a single individual. Even more important, most different
single chromosome hybrid cell lines have |
been made from different individuals. So the |
||
real answer to the often asked question “Who will be sequenced in the human genome |
|||
project” is that the sequence will inevitably represent a mosaic of many individuals. This |
|||
is probably quite appropriate, given the global nature and implications of the project. |
|||
There are other bars to total genome sequencing, or even total sequencing of a given |
|||
chromosome. We have indicated many times already that closure in mapping is a difficult |
|||
task; closure in large-scale sequencing projects will also be extremely difficult. For what- |
|||
ever reason, there are bound to be a few regions in any chromosome that cannot be cloned |
|||
by any of |
our existing methods, or that may not be approachable even by PCR or ge- |
||
nomic sequencing. Sequences with very peculiar secondary structures or sequences toxic |
|||
to the enzymes or cells we must rely on |
could lead to this kind of problem. The issue of |
||
how much effort should be devoted to a |
few missing stretches has not yet been forced |
||
upon us, but placed in any kind of reasonable perspective, it cannot have high priority rel- |
|||
ative to more productive use of large-scale sequencing facilities. |
|||
Finally, some regions of chromosomes are either very variable or dull, at least at the |
|||
level of fine details. Examples are long simple sequence or tandemly repeating sequence |
|||
repeats. Human centromeres appear to have millions of base pairs of such repeats. Other |
|||
heterochromatic regions are occasionally seen on certain chromosomes. Some of these re- |
|||
gions show quite significant size variation within the human population. For example, a |
|||
case is known of |
an apparently healthy individual with one copy of chromosome 21 that |
||
is 50% longer than average. Surely we will not select these extra long variants for initial |
|||
mapping or sequencing projects. However, the key point is that extensive, large-scale se- |
|||
quencing of simple repeats does not seem to be justified at the present time by any hints |
|||
that this large amount of sequencing data will be interesting or interpretable. Furthermore |
|||
our current methods are actually incapable of dealing with such regions of the genome. |
|||
Thus we will almost certainly have to claim completeness, missing the centromeres and |
|||
certain other unmanageable genome regions. |
|
||
SEQUENCING |
THE |
FIFTH BASE |
|
When we have sequenced all of the cloned DNAs from each human single chromosome |
|||
library, we will not have the complete |
DNA sequence of an individual, for the reasons |
||
cited above. Some of the troublesome regions are almost certainly not in our libraries or, |
|||
if they are present, they probably represent badly rearranged remnants of what was actu- |
|||
ally in the genome. If we ever want to |
look at such sequences in their native state, we |
||
may have to sequence them directly from |
the genome. For the reasons cited above, this |
||
may not be a terribly interesting or useful thing to do. However, there is a tremendously |
|||
important additional reason to perfect methods for direct genomic mammalian sequenc- |
|||
ing. This is to look at the fifth base, |
m C, which is lost in all common current cloning sys- |
||
tems. It is also lost in PCR amplification. Therefore, to find the location of malian or other higher eukaryotic DNA sequences, it is necessary to immortalize the positions of these residues before any amplification.
PCR can be used to determine DNA sequence directly from genomic samples with mammalian complexity by a ligation technique that is shown in Figure 11.22
mC in mam-
a. The ge-