

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
3 Analysis of DNA Sequences
by Hybridization
BASIC |
REQUIREMENTS |
FOR |
SELECTIVITY |
AND |
SENSITIVITY |
|
|
|
|
|
|
|
|
||||||||
The haploid human genome is 3 |
|
|
|
10 |
9 |
base |
pairs, and a |
typical human cell, as described in |
|||||||||||||
the last chapter, is somewhere between diploid and tetraploid in DNA content. Thus each cell |
|
|
|
|
|||||||||||||||||
has about 10 |
10 |
base pairs of DNA. A single base pair is 660 Da. Hence the weight of DNA in a |
|
|
|||||||||||||||||
single cell can be calculated as 10 |
|
|
10 |
|
660 / (6 |
10 |
23 ) 10 |
11 |
g or 10 pg. Ideally we would |
||||||||||||
like to be able to do analyses on single cells. This means that if only a small portion of the |
|
|
|||||||||||||||||||
genome is the target for analysis, far less than 10 pg of material will need to be detected. By |
|
|
|
||||||||||||||||||
current methodology we are in fact able to determine the presence or |
absence of almost |
any |
|
|
|
|
|||||||||||||||
20-bp DNA sequence within a single cell, such as the sequence ATTGGCATAGGAGCC- |
|
|
|
|
|||||||||||||||||
CATGG. This analysis takes place at the level of single molecules. Two requirements must be |
|
|
|
|
|||||||||||||||||
met to perform such an exquisitely demanding analysis. There must be sufficient experimental |
|
|
|
|
|||||||||||||||||
sensitivity to detect the presence of the sequence. This sensitivity is provided by either chemi- |
|
|
|||||||||||||||||||
cal or |
biological |
amplification |
procedures or |
by |
a combination of these procedures. |
There |
|
|
|
|
|||||||||||
must also be sufficient experimental selectivity to discriminate between the desired, true target |
|
|
|
|
|||||||||||||||||
sequence and all other similar sequences, which may differ from the target by as little as one |
|
|
|
||||||||||||||||||
base. That specificity lies with the intrinsic selectivity of DNA base pairing, itself. |
|
|
|
|
|
|
|
||||||||||||||
The target of a 20-bp DNA sequence is not picked casually. Twenty bp is just about the |
|
|
|||||||||||||||||||
smallest DNA length that has a high probability, |
a priori, of being found in a |
single copy |
|
|
|
||||||||||||||||
in the human genome. This can be deduced as |
follows |
from |
simple binomial |
statistics |
|
|
|
|
|||||||||||||
(Box 3.1). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
For |
simplicity, |
pretend |
that the human genome contains equal amounts |
of |
the |
four |
|
|
|||||||||||||
bases, A, T, C, and G, and |
that the occurrences of the bases are random. (These con- |
|
|
||||||||||||||||||
straints will be relaxed elsewhere in the book when some of the unusual statistical proper- |
|
|
|
||||||||||||||||||
ties of natural DNAs need to be considered explicitly. Then the expected frequency of oc- |
|
|
|
||||||||||||||||||
currence |
of any |
particular |
stretch |
of |
DNA |
sequence, |
|
such |
as |
|
|
|
|
|
|
n |
bases beginning as |
||||
ATCCG . . ., is 4 |
|
n . The |
average number |
|
of |
occurrences |
of this particular sequence in |
|
|||||||||||||
the haploid human genome is 3 |
|
|
|
10 |
9 |
4 n . For a sequence of 16 bases, |
n 16, the av- |
||||||||||||||
erage |
occurrence |
is 3 |
|
10 |
9 4 16 |
which is |
about 1. Thus |
such a |
length |
will tend to |
be |
||||||||||
seen as often as not by chance; it is not long enough to be a unique identifier. There is a |
|
|
|||||||||||||||||||
reasonable chance that the sequence 16 bases long will occur several times in different |
|
|
|||||||||||||||||||
places in the genome. Choosing |
|
|
|
n |
20 gives an average occurrence of about 0.3%. Such |
||||||||||||||||
sequences will almost always be unique genome landmarks. One corollary of this simple |
|
|
|
|
|||||||||||||||||
exercise is that it is a very futile exercise to look at random for the occurrence of particu- |
|
|
|||||||||||||||||||
lar 20-mers in the sequence of a higher organism unless there is good a priori reason for |
|
|
|||||||||||||||||||
suspecting the presence of these sequences. This means |
that sequences of |
length 20 |
or |
|
|
||||||||||||||||
more can be used as unique identifiers (see Box 3.2). |
|
|
|
|
|
|
|
|
|
|
|
||||||||||
64
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BASIC REQUIREMENTS FOR SELECTIVITY AND SENSITIVITY |
|
|
65 |
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
BOX 3.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
BINOMIAL |
|
STATISTICS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
Binomial |
|
statistics |
describe |
the |
probable outcome of events like coin |
flipping, |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
events that depend on a |
single random variable. While a normal coin has a |
|
50% |
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
chance of heads or tails |
with each flip, we will consider here the more general |
case |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
of |
|
a |
weighted |
|
coin |
with |
two |
possible |
outcomes |
with |
probabilities |
|
|
|
|
|
|
|
|
|
p |
(heads) |
and |
|
||||||||||||||||
q |
|
(tails). Since there are no other possible outcomes |
|
|
|
|
|
|
p |
q |
1. If N |
successive flips |
|
|
||||||||||||||||||||||||||
are executed, and the outcome is a particular string, such as |
|
|
|
|
|
|
|
|
hhhhttthhh, |
the |
chance |
|
||||||||||||||||||||||||||||
of this particular outcome is |
|
|
|
p n q N n , |
where |
|
n |
is the number |
of times heads was ob- |
|
|
|
|
|||||||||||||||||||||||||||
served. Note that all strings with the same numbers of heads and tails will have the |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
same a priori probability, since in binomial statistics each event does not affect the |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
probability of subsequent events. Later in this book we will deal with cases where |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
this extremely simple model does not hold. If we care only about the chance of an |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
outcome |
with |
|
|
|
|
|
|
n |
heads |
and |
|
N |
n |
tails, |
without regard |
to |
sequence, |
the |
number of |
|
|
|
|
|||||||||||||||||
such |
events |
is |
|
|
|
|
|
N |
!/(n !)(N n )!, and so the fraction of |
times |
this |
outcome |
will |
be |
|
|
|
|
||||||||||||||||||||||
seen is ( |
|
p n q N n )N !/(n !)(N n )! |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
A simple binomial model can also be used to estimate the frequency of |
occur- |
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
rence |
of |
particular |
DNA |
base sequences. Here there are four possible outcomes |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
(not quite as complex as dice throwing where six possible outcomes occur). For a |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
particular |
string |
|
with |
|
|
|
n A A’s, |
n C |
C’s, |
n G |
G’s and |
n T |
T’s, and |
a base |
|
composition |
of |
n T . |
|
|
||||||||||||||||||||
X |
A |
, |
X |
C |
, |
X |
|
G |
, |
and |
|
X |
T |
the |
chance occurrence |
of |
that |
string |
is |
|
|
|
|
|
X |
|
n A X |
n C X |
n G X |
The |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
C |
G |
T |
|
|
|||||
number of possible strings with a particular base composition is |
|
|
|
|
|
|
|
|
|
N |
!/(n A |
!n C !n G !n T !), |
|
|||||||||||||||||||||||||||
and by combining this with the previous term, the probability of a string with a par- |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
ticular base composition can easily be computed. Incidentally, the number of possi- |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
ble strings |
of |
length |
|
|
|
|
N |
is |
4 N |
, while |
|
the |
number |
of different |
base |
compositions |
|
of |
|
|
|
|
|
|||||||||||||||||
this length is ( |
|
|
|
N |
|
3)!/(N !3!). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
The |
same |
|
statistical |
models can |
be used to make estimates that two people |
will |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
share the same DNA sequences. Such estimates are very useful in DNA-based identity |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
testing. Here we consider just the simple case of two allele polymorphisms. In a par- |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
ticular place in the genome, suppose that a fraction of all individuals have one base, |
|
|
|
|
|
|
|
|
f1 , |
|
||||||||||||||||||||||||||||||
while the remainder have another, |
|
|
|
|
f2. The chance that two individuals share |
the |
same |
|
|
|
|
|
||||||||||||||||||||||||||||
allele is |
|
f |
1 |
2 f |
2 |
2 |
g |
2. If |
a set |
of |
M |
|
two-allele |
polymorphisms |
( |
|
|
|
i, j, k, . . .) |
is consid- |
|
|||||||||||||||||||
ered |
simultaneously, |
the |
chance |
that |
two |
individuals |
are |
identical |
for |
all |
of |
them is |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
g |
2i g j2g k2. . |
|
. . |
By |
|
choosing |
|
M |
sufficiently |
large, |
we |
can |
clearly |
make |
the |
overall |
|
|
|
|
|
|||||||||||||||||||
chance too low to occur, |
unless the individuals in question are one and the |
same. |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
However, two caveats apply to this |
reasoning. First, related individuals will |
show |
a |
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
much higher degree of similarity than predicted by this model. Monozygotic twins, in |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
principle, should share an identical set of alleles at the germ-line level. Second, the |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
proper allele frequencies to use will depend on the racial, ethnic, and other genetic |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
characteristics of the individuals in question. Thus it may not always be easy to select |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
appropriate values. These difficulties notwithstanding, DNA testing offers a very pow- |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
erful approach to identification of individuals, paternity testing, and a variety of foren- |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
sic applications. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
66 |
ANALYSIS OF DNA SEQUENCES BY |
HYBRIDIZATION |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
BOX 3.2 |
|
|
|
|
|
|
|
|
|
|
|
DNA SEQUENCES |
AS |
UNIQUE SAMPLE IDENTIFIERS |
|
|
|
|
|
|
|||
The following table shows the number of different |
sequences |
of length |
|
n |
and com- |
|
|||||
pares these values to the sizes of various genomes. Since genome size is virtually the |
|
|
|
|
|||||||
same as the number of possible short substrings, it is easy to determine the lengths of |
|
|
|
|
|||||||
short sequences that will occur on average only |
once per genome. Sequences a few |
|
|
|
|
||||||
bases longer than these lengths will, for all practical purposes, occur either once or not |
|
|
|
|
|||||||
at all, and hence they can serve as unique identifiers. |
|
|
|
|
|
||||||
LENGTH |
|
NUMBER OF SEQUENCES |
GENOME |
, GENOME SIZE (BP ) |
|
||||||
|
8 |
|
6.55 |
10 |
4 |
Bacteriophage lambda, 5 |
|
10 4 |
|
||
|
9 |
|
2.60 |
|
10 |
5 |
|
|
|
|
|
|
10 |
|
1.05 |
|
10 |
6 |
|
|
|
|
|
|
11 |
|
4.20 |
|
10 |
6 |
E. coli, |
4 10 6 |
|
|
|
|
12 |
|
1.68 |
|
10 |
7 |
S. cerevisiae, 1.3 |
10 |
7 |
|
|
|
13 |
|
6.71 |
|
10 |
7 |
|
|
|
|
|
|
14 |
|
2.68 |
10 |
8 |
All mammalian mRNAs, 2 |
|
10 |
8 |
||
|
15 |
|
1.07 |
|
10 |
9 |
|
|
|
|
|
|
16 |
|
4.29 |
|
10 |
9 |
Human haploid genome, 3 |
|
10 |
9 |
|
|
17 |
|
1.72 |
|
10 |
10 |
|
|
|
|
|
|
18 |
|
6.87 |
10 |
10 |
|
|
|
|
|
|
|
19 |
|
2.75 |
|
10 |
11 |
|
|
|
|
|
|
20 |
|
1.10 |
|
10 |
12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
DETECTION OF SPECIFIC DNA SEQUENCES |
|
|
|
|
|
|
|
||||
DNA molecules themselves are the perfect set of reagents to identify particular DNA se- |
|
|
|
|
|||||||
quences. This is because of the strong, sequence-specific base pairing between comple- |
|
|
|
|
|||||||
mentary DNA strands. Here one strand of DNA will be considered to be a target, and the |
|
|
|
|
|||||||
other, a probe. (If both are not initially available in a single-stranded form, there are many |
|
|
|
|
|||||||
ways to circumvent this complication.) The analysis for a particular DNA sequence con- |
|
|
|
|
|||||||
sists in asking whether a probe can find its target |
in the sample of interest. If the probe |
|
|
|
|
||||||
does so, a double-stranded DNA complex will be formed. This |
process is called |
|
|
|
hy- |
||||||
bridization, |
and all we have to do is to discriminate between this complex and the initial |
|
|
|
|
||||||
single-stranded starting materials (Fig. 3.1 |
|
|
|
a ). |
|
|
|
|
|||
|
The earliest hybridization experiments were carried out in homogeneous solutions. |
|
|
|
|
||||||
Hybridization was allowed to proceed for a fixed time period, and then a physical separa- |
|
|
|
|
|||||||
tion was performed to capture double-stranded material and discard single strands. |
|
|
|
|
|||||||
Hydroxyapatite chromatography was used to do this |
discrimination because conditions |
|
|
|
|
||||||
could be found in which double-stranded DNA bound |
to a column |
of hydroxylapatite, |
|
|
|
|
|||||
while single strands were eluted (Fig. 3.1 |
|
|
|
b). The amount of double-stranded DNA could |
|
|
|
||||
be quantitated by using a radioisotopic label on the |
probe or the target, or by measuring |
|
|
|
|
||||||
the |
bulk amount |
of |
DNA captured or eluted. |
This method is still used today in select |
|
|
|
|
|||
cases, but it is very tedious because only a few samples can be conveniently analyzed simultaneously.
EQUILIBRIA BETWEEN DNA DOUBLE AND SINGLE STRANDS
Figure 3.1 Detecting the formation of specific double-stranded DNA sequences.
is to tell whether the sequence of interest is present in single-stranded (s.s.) or double-stranded (d.s.) form. (b)Physical purification by methods such as hydroxyapatite chromatography. Physical purification by the use of one strand attached to an immobilized phase. A label is used to
detect hybridization of the single-stranded probe.
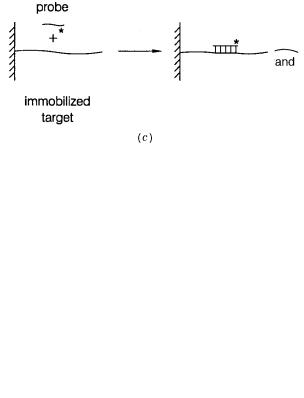
Modern hybridization protocols immobilize one of the two DNAs on a solid support (Fig. 3.1 c ). The immobilized phase can be either the probe or the target. The complementary sample is labeled with a radioisotope, a fluorescent dye, or some other specific moiety that later allows a signal to be generated in situ. The amount of color, or radioactivity, on the immobilized phase is measured after hybridization, for a fixed period, and subsequent washing of the solid support to remove adsorbed, but nonhybridized, material. As
will be shown later, an advantage of this method is that many samples can be processed in parallel. However, there are also some disadvantages that will become apparent as we proceed.
EQUILIBRIA BETWEEN DNA |
DOUBLE |
AND |
SINGLE |
STRANDS |
|
|
The |
fraction of singleand double-stranded DNA in solution can be monitored by vari- |
|||||
ous |
spectroscopic properties that effectively average over different |
DNA sequences. |
||||
Such measurements allow |
us to view |
the |
overall |
reaction of DNA |
single strands. |
|
67
(a)The problem
(c)
68 |
ANALYSIS OF DNA SEQUENCES BY HYBRIDIZATION |
|
|
|
|||
Ultraviolet absorbance, circular dichroism, or the fluorescence of dyes that bind selec- |
|
||||||
tively to duplex DNA can all be used for this purpose. If the amount of double-stranded |
|
||||||
(duplex) DNA in a sample is monitored as a function of temperature, the results typi- |
|
||||||
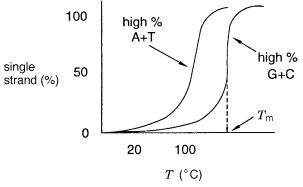
cally obtained are shown in Figure 3.2. The DNA is transformed from double strands at |
|
|
|||||
low temperature, rather abruptly at some critical temperature, to single strands. The |
|
||||||
process, for long DNA, is |
usually so cooperative that it |
can |
be likened to |
the melting |
of |
|
|
a solid, and the transition is called |
DNA |
melting. |
The midpoint of |
the transition for a |
|||
particular DNA sample is called the melting temperature, |
|
|
T m . For DNAs that are very |
||||
rich in the bases G |
C, this can be 30 or 40°C higher than for extremely (A |
T)-rich |
|||||
samples. It is such spectroscopic observations, on large numbers of |
small DNA |
du- |
|
||||
plexes that have allowed us to achieve a quantitative |
understanding of |
most aspects |
of |
|
|||
DNA melting. |
|
|
|
|
|
|
|
Figure 3.2 |
Typical melting |
behavior for DNA as a function of average base composition. Shown |
|
||||||||||
is the fraction of single-stranded molecules as a function of temperature. The midpoint of the transi- |
|
||||||||||||
tion is the melting temperature, |
|
|
|
T |
m . |
|
|
|
|
|
|
||
The goal of this section is to define conditions that allow sequence-specific analyses of |
|
||||||||||||
DNA using DNA hybridization. Specificity means the ratio of perfectly base-paired du- |
|
|
|||||||||||
plexes to duplexes with imperfections |
or |
mismatches. Thus |
high |
specificity means |
that |
|
|
||||||
the conditions maximize the amount |
of double-stranded perfectly base-paired complex |
|
|
||||||||||
and minimize the amount of other species. Key |
variables are |
the concentrations of |
the |
|
|||||||||
DNA probes and targets that are used, the temperature, and the salt concentration (ionic |
|
||||||||||||
strength). |
|
|
|
|
|
|
|
|
|
|
|
|
|
The melting temperature of long |
DNA is concentration |
|
|
|
in dependent. This arises from |
|
|||||||
the way in |
which |
T m |
is |
defined. Large DNA melts in patches as shown in |
Figure 3.3 |
a . At |
|||||||
T m , the temperature at which half |
the |
DNA |
is |
melted, |
(A |
|
|
T)-rich zones are melted, |
|
||||
while (G |
C)-rich zones |
are still in duplexes. No net strand separation will have taken |
|
||||||||||
place because no duplexes will have been completely melted. Thus there can be no con- |
|
|
|||||||||||
centration dependence to T |
|
|
|
m . |
|
|
|
|
|
|
|
||
In contrast, the melting of short DNA duplexes, DNAs of 20 base pairs or less, is ef- |
|
||||||||||||
fectively all or none (Fig. 3.3 |
|
|
|
b). In this case the concentration of intermediate species, |
|
||||||||
partly single-stranded and partly duplex, is sufficiently small that it can be ignored. The |
|
||||||||||||
reaction of two short complementary DNA strands, |
|
|
|
A |
and |
B, may be written as |
|
||||||
A B AB

EQUILIBRIA BETWEEN DNA DOUBLE AND SINGLE STRANDS |
69 |
Figure 3.3 |
Melting behavior of DNA. |
(a)Structure of a typical high molecular weight DNA at its |
melting temperature. (b)Status of a short DNA sample at its melting temperature.
The equilibrium (association) constant for this reaction is defined as
K a [AB ] [A ][B ]
Most experiments will start with an equal concentration of the two strands (or a duplex melted to give the two strands). The initial total concentration of strands (whatever their
form) is |
C |
T |
. If, for simplicity, all of the strands are initially single stranded, their concen- |
|||||||||||||||||||||||||
trations is |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[A ] |
|
[B ] |
C |
T |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
o |
|
|
|
o |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
At |
T m |
half |
|
the |
strands must be |
in |
duplex. |
Hence |
the |
concentrations |
|
of |
|
the |
|
different |
||||||||||||
species at |
|
T |
m |
will be |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[AB ] |
[A ] [B ] |
|
|
C |
T |
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
The equilibrium constant at |
|
T |
m |
is |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
K |
a |
|
[AB |
] |
|
|
C |
T |
/ 4 |
|
|
|
4 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
[A ][B ] (C T / 4) C T |
|||||||||||||||||
Do not be misled by this expression into thinking that the equilibrium constant is concen- |
||||||||||||||||||||||||||||
tration dependent. It is the |
T |
m |
that is concentration dependent. The equilibrium constant is |
|||||||||||||||||||||||||
temperature dependent. The above expression indicates the value seen for the equilibrium |
|
|
|
|
||||||||||||||||||||||||
constant, 4/ |
|
C |
T , at the temperature, |
|
|
|
|
T m |
. This particular |
|
|
|
|
|
T |
m occurs when the equilibrium is |
||||||||||||
observed at the total strand concentration, |
|
|
|
|
|
|
C |
T . |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
70 |
ANALYSIS OF DNA SEQUENCES BY HYBRIDIZATION |
|
|
|
|
|
|
|
|
|
|
||||||||||||
A special case must be considered in which hybridization occurs between two short |
|
||||||||||||||||||||||
single |
strands |
of the same |
identical |
sequence, |
|
|
|
|
|
|
|
C. |
|
Such strands are self-complementary. |
|||||||||
An example is GGGCCC which can base pair with itself. In this case the reaction can be |
|
||||||||||||||||||||||
written |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2C |
C |
2 |
|
|
|
|
|
|
|
|
|
||||
The equilibrium constant, |
|
|
K a , becomes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
K a |
|
[C |
2] |
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
[C |
2 |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
] |
|
|
|
|
|
|
|
|
|
|||
At the melting temperature half of the strands must be duplex. Hence |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
[C 2] |
[2C ] |
|
C T |
|
|
|
||||||||||||
|
|
|
|
|
4 |
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
where |
C T |
as before is the total concentration of strands. Thus we can evaluate the equilib- |
|
||||||||||||||||||||
rium expression at |
T m |
as |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
K |
a |
C T |
/ 4 |
|
|
|
1 |
|
|
|
|
||||||
|
|
|
|
|
|
(C T / 2) |
C |
|
|
T |
|
|
|||||||||||
As before, what this really means is that |
|
|
|
|
T m |
|
is |
concentration dependent. |
In both cases |
||||||||||||||
simple mass action considerations ensure that |
|
|
|
|
|
|
|
|
T m |
|
|
will increase as the concentration is |
|||||||||||
raised. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The final case we need to consider is when one strand is in vast excess over the other |
|
||||||||||||||||||||||
instead of both being at equal concentrations. This is frequently the case |
|
|
when a |
trace |
|
||||||||||||||||||
amount of probe is used to interrogate a concentrated |
sample or, alternatively, when |
a |
|
||||||||||||||||||||
large amount of probe is used to interrogate a very minute sample. The formation of du- |
|
|
|||||||||||||||||||||
plex can be written as before as |
|
A B |
AB |
, but now the initial starting conditions are |
|||||||||||||||||||
|
|
|
|
|
|
|
|
[B ] [A ] |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
o |
|
|
|
|
o |
|
|
|
|
|
|
|
||
Effectively the total strand concentration, |
|
|
|
|
C T |
, is thus simply |
the initial |
concentration of |
|||||||||||||||
the excess strand: |
B o . At |
T m , |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
[AB |
] [A ] |
|
|
|
|
|
|
|
|||||||
Thus the equilibrium expression at |
T |
m |
becomes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
K |
a |
|
[AB |
] |
|
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
C T |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
[A ][B ] |
|
|
|
|
||||||||||
The melting temperature is still concentration dependent. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
The importance of our ability to drive duplex formation cannot be |
underestimated. |
|
|||||||||||||||||||||
Figure 3.4 illustrates the practical utility of this ability. It shows the concentration depen- |
|
||||||||||||||||||||||
dence |
of the melting temperature |
of two different duplexes. We can characterize |
each re- |
|
|
||||||||||||||||||

THERMODYNAMICS OF THE MELTING OF SHORT DUPLEXES |
71 |
Figure 3.4 |
The dependence of the |
melting temperature, |
T m , of two short duplexes on the total |
concentration of DNA strands, |
C T . |
|
|
action by its melting temperature and can attempt to extract thermodynamic parameters |
|
|
|
|
||||||||
like |
the |
enthalpy |
change, |
|
H , and the free energy change, |
G |
, for |
each |
reaction. |
|||
However, with the |
|
T m ’s |
different for the two reactions, if we do this at |
|
T |
m , the |
thermody- |
|||||
namic parameters derived will refer to reactions at two different temperatures. There will |
|
|
|
|
||||||||
be no way, in general, to compare these parameters, since they are expected to be intrinsi- |
|
|
|
|
||||||||
cally temperature dependent. The concentration dependence of melting saves us from this |
|
|
|
|
||||||||
dilemma. By varying the concentration, we can produce conditions where the |
two du- |
|
|
|
|
|||||||
plexes |
have |
the same |
T |
m . Now thermodynamic parameters derived from each are |
compa- |
|
|
|
||||
rable. We can, if we wish, choose any temperature for this comparison. In practice, 298 K |
|
|
|
|||||||||
has been chosen for this purpose. |
|
|
|
|
|
|
|
|
||||
THERMODYNAMICS OF THE MELTING OF SHORT DUPLEXES |
|
|
|
|
|
|||||||
The model we will use to analyze the melting of short DNA double helices is shown in |
|
|
|
|
||||||||
Figure 3.5. The two strands come |
together, in a nucleation step, to form a single pair. |
|
|
|
||||||||
Double strands can form by stacking of adjacent base pairs above or below the initial nu- |
|
|
|
|
||||||||
cleus until a full duplex has zippered up. It does not matter, in our treatment, where the |
|
|
|
|||||||||
initial nucleus forms. We also need |
not consider any intermediate steps beyond the nu- |
|
|
|
|
|||||||
cleus |
and the fully |
duplex state. In |
that state, for a duplex of |
|
|
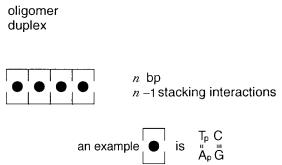
n base |
pairs, |
there will be |
||||
n 1 stacking |
interactions (Fig. |
3.6). Each interaction reflects |
the |
energetics |
of stacking |
|
|
|
||||
two adjacent base |
pairs on top of |
each other. There are ten distinct such |
interactions, |
as |
|
|
|
|||||
ApG/CpT, ApA/TpT, and so on (where the slash indicates two complementary antiparal-
lel strands). Because their energetics are very different, we must consider the DNA sequence explicitly in calculating the thermodynamics of DNA melting.
Figure 3.5 A model for the mechanism of the formation of duplex DNA from separated complementary single strands.
72 ANALYSIS OF DNA SEQUENCES BY HYBRIDIZATION
Figure 3.6 Stacking interactions in double-stranded DNA.
For each of the ten possible stacking interactions, we can define a standard |
|
|
for the |
|
|
|
|||||||||||||
free energy of |
stacking, a |
0 |
|
of |
stacking, |
and |
a |
|
for |
the entropy |
of |
S |
0 |
||||||
for the enthalpyH |
|
s |
|||||||||||||||||
|
|
|
|
|
s |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
stacking. These quantities will be related at a particular temperature by the expression |
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
G s0 |
H s0 T S s0 |
|
|
|
||||||||
For any particular duplex DNA sequence, we can compute the |
thermodynamic parame- |
|
|
|
|
||||||||||||||
ters for duplex formation by simply combining the parameters for the competent stacking |
|
|
|
|
|||||||||||||||
interactions plus a nucleation term. Thus |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
G |
0 |
|
G |
nuc0 |
G |
s0 |
|
g sym |
|
|
|
|||
|
|
|
|
|
|
|
H |
0 |
H |
nuc0 |
H |
s0 |
|
|
|
||||
and similarly |
for the entropy, where the sums are taken over all of the stacking interac- |
|
|
|
|||||||||||||||
tions in |
the |
duplex, where |
g sym |
0.4 |
kcal/mol if |
the |
two strands are identical; other- |
||||||||||||
wise, |
g sym |
0. The equilibrium constant for duplex formation is given by |
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
K ks 1 s 2s 3 . . . s n 1 |
|
|
|
|
||||||||
where |
k is the equilibrium constant of nucleation, related to the |
|
|
by |
|
G |
nuc0 |
|
|||||||||||
|
|
|
|
|
|
|
G |
nuc0 |
|
RT |
ln |
k |
|
|
|
|
|||
and each |
|
s i |
is the microscopic equilibrium |
constant |
for |
a |
particular stacking |
reaction, |
re- |
|
|
||||||||
lated to the |
|
forG |
0that reaction by |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
G |
i0 |
RT |
ln s i |
|
|
|
|
|||||
The key factor involved in predicting the stability of DNA (and RNA) duplexes is that |
|
|
|
||||||||||||||||
all of these thermodynamic parameters have been measured experimentally. One takes |
|
|
|
||||||||||||||||
advantage |
of |
the enormous power available to synthesize particular DNA sequences, |
|
|
|
||||||||||||||
combines complementary pairs of such sequences, and measures their extent of duplex as |
|
|
|
|
|||||||||||||||
a function of |
temperature and |
concentration. For |
example, we can |
study the |
properties |
of |
|
|
|
||||||||||
G
0 s
THERMODYNAMICS OF THE MELTING OF SHORT DUPLEXES
A |
/T |
8 |
and compare |
these with A |
9 |
/T |
9 |
. Since |
the only difference between these complexes |
|
|
|||||||||||
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
is an extra ApA/TpT stacking interaction, the differences will yield the thermodynamic |
|
|
|
|||||||||||||||||||
parameters for that interaction. Other sequences are more complex to handle, but this has |
|
|
|
|||||||||||||||||||
all been accomplished. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
It is helpful to choose a single standard temperature and set of environmental conditions |
|
|
|
||||||||||||||||||
for the tabulation of thermodynamic data: 298 K has been selected, and |
|
|
|
at 298 K in 1 |
G s0 's |
|
|
|||||||||||||||
M NaCl are listed in Table 3.1. Enthalpy values for the stacking interactions can be obtained |
|
|
|
|||||||||||||||||||
in two ways: either by direct calorimetric measurements or by examining the temperature de- |
|
|
|
|||||||||||||||||||
pendence of the stacking interactions. |
are also listedH in0 Table's 3.1. So are |
|
, which |
S |
0 |
's |
||||||||||||||||
can |
be |
calculated from |
the |
relationship |
|
|
|
|
|
s |
0 |
. FromH |
0 |
|
0 |
|
s |
|
||||
|
|
|
|
G |
|
|
|
|
||||||||||||||
|
|
|
|
s |
s |
these data,T Sthermody- |
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
s |
|
|
|
||
namic values at other temperatures can be estimated as shown in Box 3.3. The effects of salt |
|
|
|
|||||||||||||||||||
are well understood and are described in detail elsewhere (Cantor and Schimmel, 1980). |
|
|
|
|||||||||||||||||||
|
The results shown in Table 3.1 make |
it clear |
that |
|
the |
effects of the DNA |
sequence |
|
|
|
||||||||||||
on |
duplex stability are very large. The average |
|
|
|
is |
|
|
|
H |
|
s0 |
8 kcal/mol; the range is |
|
|
||||||||
to |
11.9 kcal/mol. The average |
is |
G |
|
s0 |
1.6 |
kcal/mol |
with a range of |
0.9 |
to |
||||||||||||
kcal/mol. Thus the DNA sequence must be considered explicitly in estimating duplex |
|
|
|
|||||||||||||||||||
stabilities. The two additional parameters needed to do this concern the energetics of nu- |
|
|
|
|||||||||||||||||||
cleation. These are relatively sequence |
independent, |
and we |
can use |
average |
|
values of |
|
|
|
|||||||||||||
G nuc0 |
|
5 |
kcal/mol (except if |
no G–C |
pairs |
are |
present, |
then |
|
|
6 |
kcal/mol should |
|
be |
||||||||
used) and |
H |
nuc0 |
0. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
For estimating the stability of perfectly paired duplexes, |
these |
nucleation |
parameters |
|
|
|
|||||||||||||||
and the stacking energies in Table 3.1 are used. Table 3.2 shows typical results when cal- |
|
|
|
|||||||||||||||||||
culated |
|
and |
experimentally |
measured |
are |
compared. TheG |
0agreement's in almost all |
|
|
|
||||||||||||
cases is excellent, and the few discrepancies seen are probably within the range of typical |
|
|
|
|||||||||||||||||||
experimental errors. The approach described above has been generalized to predict the |
|
|
|
|||||||||||||||||||
thermodynamic properties of triple helices, and presumably it will also serve for four- |
|
|
|
|||||||||||||||||||
stranded DNA structures. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
73
5.6
3.6
TABLE 3.1 |
Nearest-neighbor Stacking Interactions in |
|
|
Double-stranded DNA |
|
|
|
|
|
|
|
|
Nearest-neighbor Thermodynamics |
|
|
|
|
|
|
|
H ° |
S ° |
G ° |
Interaction |
(kcal/mol) |
(cal/Kmol) |
(kcal/mol) |
|
|
|
|
AA/TT |
9.1 |
24.0 |
1.9 |
AT/TA |
8.6 |
23.9 |
1.5 |
TA/AT |
6.0 |
16.9 |
0.9 |
CA/GT |
5.8 |
12.9 |
1.9 |
GT/CA |
6.5 |
17.3 |
1.3 |
CT/GA |
7.8 |
20.8 |
1.6 |
GA/CT |
5.6 |
13.5 |
1.6 |
CG/GC |
11.9 |
27.8 |
3.6 |
GC/CG |
11.1 |
26.7 |
3.1 |
GG/CC |
11.0 |
26.6 |
3.1 |
|
|
|
|
Source: Adapted from Breslauer et al. (1986).