

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
7 Cytogenetics and Pseudogenetics
WHY GENETICS IS INSUFFICIENT
The previous chapter showed the potential power of linkage analysis in locating human disease genes and other inherited traits. However, as demonstrated in the chapter, genetic analysis by current methods is extremely tedious and inefficient. Genetics is the court of
last |
resort |
when |
no |
more direct way to locate a |
gene of interest is available. Frequently |
one |
already |
has |
a |
DNA marker believed to be |
of interest in the search for a particular |
gene, or just of interest as a potential tool for higher-resolution genome mapping. In such cases it is almost always useful to pinpoint the approximate location of this marker within
the genome as rapidly and efficiently as possible. This is the realm of cytogenetic and pseudogenetic methods. While some of these have potentially very high resolution, most
often they are used at relatively low resolution to provide the first evidence for the chro-
mosomal |
and subchromosomal location of a DNA marker of interest. Unlike genetics, |
which can work with just a phenotype, most cytogenetics and pseudogenetics are best ac- |
|
complished by direct hybridization using a cloned DNA probe or by PCR. |
|
SOMATIC |
CELL GENETICS |
In Chapter 2 we described the availability of human-rodent hybrid cells that maintain the full complement of mouse or hamster chromosomes but lose most of their human complement. Such cells provide a resource for assigning the chromosomal location of DNA
probes or PCR products. In the simplest |
case a panel of 24 cell lines can be used, each |
||
one containing |
only a single human chromosome. In practice, it is more efficient to use |
||
cell |
lines that |
contain pools of human chromosomes. Then simple binary logic (presence |
|
or absence of a signal) allows the chromosomal location of a probe to be inferred with a |
|||
smaller number |
of hybridization experiments or PCR amplifications. In Chapter 9 some |
||
of the principles for constructing pools will be described. However, the general power of |
|||
this approach is indicated by the example in Figure 7.1. The major disadvantage of using |
|||
pools |
is that |
they are prone to errors if |
the probe in question is not a true single-copy |
DNA |
sequence |
but, in fact, derives from a |
gene family with representatives present on |
more than one human chromosome. In this case the apparent assignment derived from the
use of pools may be totally erroneous. A second problem with the use of hybrid cells, in general, is the possibility that a given human DNA probe might cross-hybridize with rodent sequences. For this reason it is preferable to measure the hybridization of the probe not by a dot blot but by a Southern blot after a suitable restriction enzyme digestion. The reason for this is that rodents and humans are sufficiently diverged that although a similar DNA sequence may be present, there is a good chance that it will lie on a different-sized DNA restriction fragment. This may allow the true human-specific hybridization to be distinguished from the rodent cross-hybridization.
208
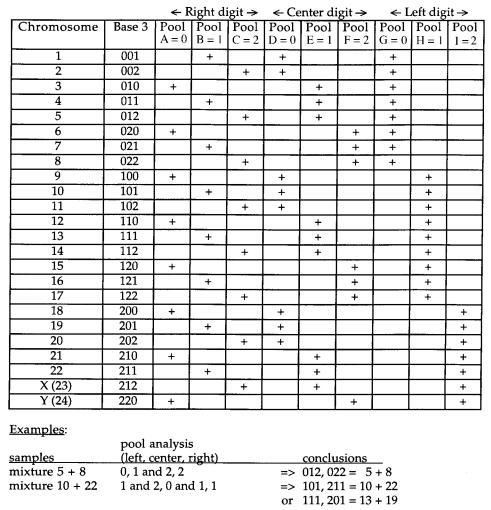
SOMATIC CELL GENETICS |
209 |
Figure 7.1 Assignment of a cloned DNA probe to a chromosome by hybridization against a panel of cell lines containing limited sets of human chromosomes in a rodent background. This hypotheti-
cal example is based on a base three pooling (see Chapter 9) which is quite efficient compared with randomly selected pools. If the probe is derived from a single chromosome, it can be uniquely assigned. If it contains material that hybridizes to two or more chromosomes, in some cases all of the chromosomes involved can be identified, but in most cases the results will be ambiguous as shown
by the example at the bottom of the figure.
An alternative way to circumvent the problem of rodent cross-hybridization in assigning DNA probes to chromosomes is to use flow-sorted human chromosomes, preferably chromosomes that have been sorted from human-rodent hybrid cells so that the purified
human fraction has very little contamination from other human chromosomes (Chapter
2). This is a very powerful approach; its major limitation is the current scarcity of flowsorted human chromosomal DNA.

210 CYTOGENETICS AND PSEUDOGENETICS
SUBCHROMOSOMAL MAPPING PANELS
Frequently it is possible to assemble sets of cell lines containing fragments of only one chromosome of interest. The occurrences of a DNA probe sequence in a subset of these cell lines can be used to assign the probe to a specific region of the chromosome. The accuracy of that assignment is determined by the accuracy with which the particular chromosome fragments contained in each cell line are known. This can vary quite considerably, but in general this approach is quite a powerful and popular one. A simple example is shown in Figure 7.2. Here three cell lines are used to divide a chromosome into three regions. The key aspect of these cell lines are breakpoints that determine which chromosome segments are present and which are absent.
A more complex example of cell lines suitable for regional assignment is given in Figure 7.3. Here the properties of a chromosome 21 mapping panel are illustrated. It is evident that some regions of the chromosome are divided by the use of this panel into very high resolu-
tion zones while others are defined much less precisely. In the case shown in Figure 7.3, human cell lines were assembled from available patient material: individuals with translocations or chromosomal deletions. Then these chromosomes were moved by cell fusion into
rodent lines to simplify hybridization analyses. Because no systematic method was used to assemble the panel, the resulting breakpoint distribution is very uneven.
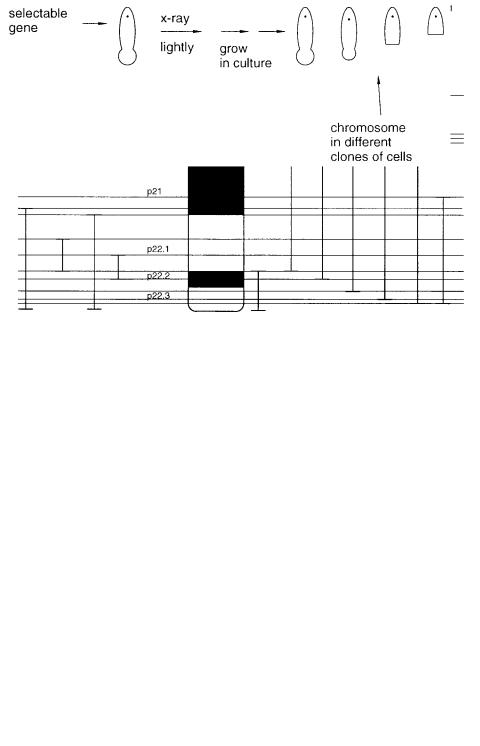
If a selectable marker exists near the end of a chromosome, it is possible to assemble a very orderly and convenient mapping panel. Assuming the selection can be applied in a
hybrid cell, what is done is to irradiate the cell line lightly with X rays so as to cause an average of less than one break per chromosome, and then grow the cells in culture and al-
low broken chromosomes to heal. Since the selection applies to only one end of the human chromosome, there will be a tendency to keep chromosomes containing that end and
lose pieces without that end. The result is a fine, ordered subdivision of the chromosome, as shown in Figure 7.4. This resource is exceptionally useful for mapping the location of cloned probes because the results are usually unambiguous. The pattern of hybridization
should be |
a step function and the point where |
positive |
signal |
disappears |
indicates the |
most distal possible location of the probe. |
|
|
|
|
|
A few caveats are in order concerning the use of cell lines that derive from broken or |
|||||
rearranged |
human chromosomes. Broken chromosome |
are always |
healed |
by fusion |
with |
other chromosomes (or by acquisition of telomeres). For example, cell lines described as 21qare missing the long arm of chromosome 21. Frequently such cell lines have other human chromosomes present that have been quietly ignored or forgotten. This is not a problem when a single-copy probe is used, assuming that this probe is already known to
Figure 7.2 A simple example of regional assignment of a DNA sample by hybridization to three cell lines containing incomplete copies of the chromosome of interest. The two break points in the chromosomes are used to divide the chromosome into three intervals.
SUBCHROMOSOMAL MAPPING PANELS |
211 |
Figure 7.3 A mapping panel of hybrid cell lines used to assign probe locations on human chromosome 21. Vertical lines indicate the human chromosome content of each cell line. Note the unevenness of the resulting intervals (horizontal lines).
be located on the chromosome of interest, and the goal is just narrowing down its subchromosomal location. However, the problem is much more serious if the probe of inter-
est comes from a gene family or contains repeated sequences. The actual chromosome content of a set of cell lines described as chromosome 21 hybrids is illustrated in Figure 7.5. Many of these lines, in fact, contain other human chromosomes. Cross-hybridization to rodent background is always a potentially serious additional complication when work-
ing with hybrid cells. For this reason it is best to do Southern blots or PCR analysis rather than dot blots, as we already explained earlier in the chapter.
Figure 7.4 A method for selection of broken chromosomes that can be used to generate a much more even mapping panel than the one shown in Figure 7.3.
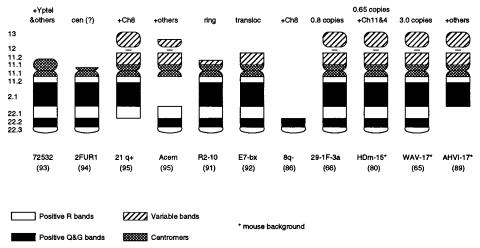
212 CYTOGENETICS AND PSEUDOGENETICS
Figure 7.5 Actual chromosome content of some of the cell lines used to produce the mapping panel summarized in Figure 7.3. Note that many contain other human chromosomes besides num-
ber 21.
RADIATION |
HYBRIDS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
It is possible to make hybrid cells containing small fragments of human chromosomes. |
|
|
|
|||||||||||||||||
These cell lines represent, essentially, a way of cloning small continuous stretches of hu- |
|
|
||||||||||||||||||
man DNA. The method was originally described by S. Goss and H. Harris, and later re- |
|
|
|
|||||||||||||||||
discovered and elaborated by D. Cox and R. Meyers (Cox et al., 1990). The basic tech- |
|
|
||||||||||||||||||
nique |
for |
producing |
these |
cells, |
called |
|
|
radiation |
hybrids, |
|
is |
shown |
schematically in |
|||||||
Figure 7.6. The starting point is a hybrid cell line containing only a single human chro- |
|
|
||||||||||||||||||
mosome in a Chinese hamster ovary (CHO) cell with a wild type hypoxanthine ribosyl |
|
|
|
|||||||||||||||||
transferase (hprt) gene. This cell line is subjected to a very high X-ray dose, 8000 rads, |
|
|
||||||||||||||||||
which is sufficient to break every chromosome into five pieces on average. After irradia- |
|
|
|
|||||||||||||||||
tion the fragments are allowed to heal, and the resulting cells are fused with an hprt |
|
|
|
CHO |
||||||||||||||||
line, one in which an inactive allele of the hprt gene is present. Cell clones that have re- |
|
|
||||||||||||||||||
ceived |
and |
retained |
hprt |
|
|
DNA are selected and maintained by growth on a particular set |
|
|||||||||||||
of |
conditions |
called |
|
|
HAT |
medium, |
|
which require an active hprt gene. Some |
of |
these |
||||||||||
clones are recipients of both CHO and human DNA. Note that no human-specific selec- |
|
|
||||||||||||||||||
tion has been used to focus |
the choice of hybrid cells on any particular region of the hu- |
|
|
|||||||||||||||||
man chromosome in question. Thus the population of hybrids should represent a random |
|
|
|
|
||||||||||||||||
sampling of that chromosome if there is no intrinsic selection method operative. |
|
|
|
|
|
|||||||||||||||
|
Selection |
for |
an |
hprt |
|
|
phenotype ensures that all |
of |
the |
cell |
lines |
maintained will |
||||||||
have |
been |
recipients |
of |
some foreign |
DNA. |
Those |
that have human DNA present |
|
|
|
||||||||||
can be detected by hybridization with human-specific |
repeated sequences. |
Then |
these |
|
|
|
||||||||||||||
lines are allowed to grow in the absence |
of any selection for human |
markers. |
This |
will |
|
|
|
|||||||||||||
lead to random loss of |
human sequences as time progresses. The |
cells |
are |
contin- |
|
|
||||||||||||||
ually screened for the presence of human markers until 30% to 60% of the hybrids con- |
|
|
||||||||||||||||||
tain |
a particular |
marker |
of |
interest. |
At this |
point |
the |
radiation hybrids contain |
quite |
|
|
|||||||||
a |
few |
discrete |
fragments of |
human |
DNA |
integrated between |
hamster |
DNA segments. |
|
|
|
|||||||||
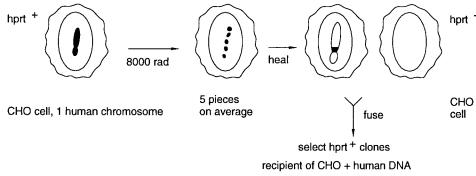
RADIATION HYBRIDS |
213 |
Figure 7.6 |
Steps in the production of a set of |
radiation hybrid cell lines. Note that there is no se- |
lection for any |
of the human DNA fragments, so they |
will be lost progressively as the cells are |
grown. |
|
|
They are now used to look at the statistics of co-retention of pairs of human DNA markers. The basic idea is illustrated below, for three markers on a chromosome:
—————A |
—— B———————————C—— |
|
|
|
|
|
|
|||
Even when the chromosome is fragmented into five pieces, there is a good chance that A |
|
|
|
|||||||
and B will still be found on the same DNA piece; it is much more likely that C will be on |
|
|
|
|||||||
a different piece. Thus cell lines that have lost A are |
also likely to have lost |
B, but they |
|
|
|
|||||
are less likely to have lost C. By establishing the presence or absence of particular mark- |
|
|
|
|||||||
ers, we gain information about the relative |
proximity |
of |
the |
markers |
because |
nearby |
|
|
|
|
markers tend to be co-retained. |
|
|
|
|
|
|
|
|
|
|
Here we will examine the statistics of co-retention of marker pairs. There is a basic |
|
|
|
|||||||
problem with this approach. If only two markers at a time are considered, it turns out that |
|
|
|
|||||||
there are more variables than constraints, and approximations have to be used to analyze |
|
|
|
|||||||
the results. With more than two simultaneous markers, the mathematics becomes more |
|
|
|
|||||||
robust but only at a cost of much greater complexity. For didactic purposes we will ana- |
|
|
|
|||||||
lyze the two-marker case; anyone with a serious interest in pursuing this further should be |
|
|
|
|||||||
aware that in practice, one must consider four or more markers at once to avoid the addi- |
|
|
|
|||||||
tional assumptions used later in this section. The parameters we must consider are illus- |
|
|
|
|||||||
trated below. The two markers A and B reside either on the same DNA fragment or on |
|
|
|
|||||||
separate fragments as shown by the horizontal dashed lines. These fragments will have |
|
|
|
|||||||
individual probabilities of retention, as indicated. |
|
|
|
|
|
|
|
|
||
————A————B———— |
retention probability |
|
|
P |
AB |
|
||||
———A——— |
——B—–— |
retention probabilities |
|
|
|
P A , P B |
|
|||
The probability that a break has occurred between A and B in a particular irradiation |
|
|
|
|||||||
treatment is given by the additional parameter |
|
|
|
. All |
four parameters |
P A , |
P B , P AB , and |
|
||
have values between 0 and 1. |
|
|
|
|
|
|
|
|
|
|
What is done experimentally is to examine |
a set of hybrid cell clones and |
to ask in |
|
|
|
|||||
each whether markers A and B are |
present. The |
fraction |
of |
them |
that have |
both markers |
|
|
|
|
214 CYTOGENETICS AND PSEUDOGENETICS
present is fAB , the fractions with only A or only B are tion that have retained neither marker is
four parameters by the following equations:
fA and fB , respectively, and the frac- f0 . These four observables can be related to the
|
fAB P AB (1 ) P A P B |
The |
first term denotes cells that have maintained A and B on an unbroken piece of DNA; |
the |
second term considers those cells that have suffered a break between A and B but |
have subsequently retained both separate DNA pieces.
fA P A (1 P B )
fB P B (1 P A )
The top equation indicates cells that have had a break between A and B and kept only A; the bottom equation indicates cells with a similar break, but these have retained only B.
|
f0 (1 P AB ) (1 ) (1 P A ) (1 P B ) |
|||||||||||
The first term indicates cells that originally contained |
A |
and |
B |
on |
a continuous |
DNA |
||||||
piece but subsequently lost this piece (probability 1 |
|
|
|
|
|
|
P AB ); the second term indicates |
|||||
cells that originally had a break between A and B and |
subsequently lost |
both |
of |
these |
||||||||
DNA pieces. |
|
|
|
|
|
|
|
|
|
|
|
|
The four types of cells represent all possibilities; |
|
|
|
|
|
|
|
|
|
|||
|
|
|
fAB fA fB f0 1 |
|
||||||||
This means that we have only three independent observables, but there are four indepen- |
|
|||||||||||
dent unknown parameters. The simplest way |
around this dilemma would |
be |
to assume |
|
|
|
||||||
that the |
probability of retention |
of |
individual markers |
was |
equal, |
for |
example, |
that |
||||
P A P |
B . However, available experimental data contradict this. Presumably some markers |
|||||||||||
lie in regions that because of chromosome structural elements like centromeres or telo- |
||||||||||||
mers, or metabolically active genes, convey a relative advantage or disadvantage for re- |
||||||||||||
tention of their region. The approximation actually used by Cox and |
Myers |
to |
allow |
|||||||||
analysis of their data is to assume that |
|
P |
A |
and |
P B |
are related |
to the total amount of A and |
|||||
B retained, respectively: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P A fAB |
fA |
|
|
|
||||
|
|
|
|
P |
B fAB |
fB |
|
|
|
|||
Upon inspection this assumption can be shown to be algebraically false, but in practice, it |
|
|||||||||||
seems to work well enough to allow sensible analysis of existing experimental data. |
|
|
|
|||||||||
The easiest way to use the two |
remaining independent |
observables |
is |
to |
perform |
the |
|
|||||
sum indicated below: |
|
|
|
|
|
|
|
|
|
|
|
|
fA fB P A (1 P B ) P B (1 P A )
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SINGLE-SPERM PCR |
215 |
This can be rearranged to yield a value for |
|
|
|
|
|
, which is the actual parameter that should be |
|
||||||||||
directly related to the inverse distance between the markers A and B: |
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
fA fB |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
P A P B 2 P A P B |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||
Here the |
parameters |
|
P |
A |
and |
P |
B are |
evaluated |
from |
measured |
values |
of |
|
fAB , fA , and |
fB as |
||
described above. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note that in using radiation hybrids, one can score any DNA markers A and B whether |
|
||||||||||||||||
they are just physical pieces of DNA or |
whether they are inherited traits that can some- |
|
|||||||||||||||
how be detected in the hybrid cell lines. |
However, |
the |
|
resulting |
radiation |
hybrid |
map, |
|
|||||||||
based on values of |
|
, is expected to be a physical map and not a genetic map. This is be- |
|
||||||||||||||
cause |
is related to the probability of X-ray breakage, which is usually considered to be |
|
|||||||||||||||
a property of the DNA itself and not in any way related to meiotic recombination events. |
|
||||||||||||||||
In most studies to date, |
|
|
has been assumed to be relatively uniform. This means that in |
|
|||||||||||||
addition |
to marker |
order, |
estimates |
for DNA |
distances |
|
between markers were inferred |
|
|||||||||
from measured values of |
|
|
. |
|
|
|
|
|
|
|
|
|
|
|
|
||
An example of a relatively mature radiation hybrid map, for the long arm of human |
|
||||||||||||||||
chromosome 21, is shown in Figure 7.7. The units of the map are in centirays (cR), where |
|
||||||||||||||||
1 cR is a distance that corresponds to 1% breakage at the X-radiation dosage used. The |
|
||||||||||||||||
density of markers on this |
map is impressively high. However, the distances in this map |
|
|
|
|||||||||||||
are not as uniform as previously thought. As shown in Table 7.1, the radiation hybrid map |
|
||||||||||||||||
is considerably elongated at both the telomeric and centromeric edges of the chromosome |
|
|
|
|
|||||||||||||
arm, relative to the true physical distances revealed by a restriction map of the chromo- |
|
||||||||||||||||
some. It is not known at present whether these discrepancies arose because of the over- |
|
||||||||||||||||
simplifications used |
to construct |
the map |
(discussed |
above) |
or |
because, |
as has |
been |
|
||||||||
shown in several studies, the probability of breakage or subsequent repair is nonuniform along the DNA. A radiation hybrid map now exists for the entire human genome. It is compared to the genetic map and to physical maps in Table 7.2.
SINGLE-SPERM PCR
A human male will never have a million progeny, but he will have an essentially unlimited number of sperm cells. Each will display the particular meiotic recombination events that generated its haploid set of chromosomes. These cells are easily sampled, but in
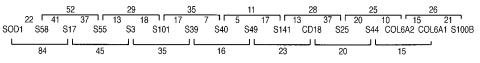
Figure 7.7 |
A radiation hybrid map of part of human chromosome 21. The units of the map are in |
|||
centirays (cR). The map shows the order of probes and the |
distance |
between |
each pair. The telo- |
|
mere is to the |
right, near S100B. Distances for adjacent |
markers |
are shown |
between the markers, |
and for nearest neighbor pairs above and below the map. (Adapted from Burmeister et al., 1994.)

TABLE 7.1 |
Comparison of the Human Chromosome 21 |
Not |
I Genomic Map |
with the Radiation Hybrid Map |
|
|
|
Marker Pairs |
Physical Distance, kb |
a |
cR b |
kb/cR a |
D21S16–D21S11, 1 |
5300 |
2400 |
87 |
36 |
16 |
||
D21S11, 1–D21S12 |
7300 |
2200 |
140 |
52 |
16 |
||
D21S12–SOD |
6065 |
2495 |
66 |
92 |
38 |
||
SOD–D21S58 |
1490 |
480 |
22 |
68 |
22 |
||
D21S58–D21S17 |
2310 |
1040 |
39 |
59 |
26 |
||
D21S17–D21S55 |
3860 |
1600 |
35 |
110 |
46 |
||
D21S55–D21S39 |
3700 |
1350 |
43 |
86 |
31 |
||
D21S39–D21S141, 25 |
3690 |
660 |
40 |
91 |
16 |
||
D21S151, 25–COL6A |
2330 |
540 |
54 |
43 |
10 |
||
|
|
|
|
|
|
|
|
Source: |
From Wang and Smith (1994). |
|
|
|
|
|
|
a The uncertainty shown for each distance is the maximum possible range. The distance given is the mean of that |
|
|
|
|
|||
range. The uncertainty shown for |
each ratio is the maximum range, considering only uncertainty in the place- |
|
|
|
|
||
ment of markers on the restriction map and ignoring any uncertainty in the radiation hybrid map. |
|
|
|
|
|||
b Taken from the data of Cox et al. (1990) and Burmeister et al. (1991). These data were obtained at 8000 rads.
TABLE 7.2 |
Characteristics of the Human Radiation Hybrid Map |
|
|
|
|
|
|
|
|
Average Relative Metrics |
|
|
|
|
|
|
|
Chromosome |
Physical Length (Mb) |
cM/cR |
|
a |
kb/cR a |
|
|
|
|
|
|
1 |
263 |
|
0.20 |
197 |
|
2 |
255 |
|
0.21 |
225 |
|
3 |
214 |
|
0.27 |
233 |
|
4 |
203 |
|
0.28 |
256 |
|
5 |
194 |
|
0.29 |
272 |
|
6 |
183 |
|
0.24 |
243 |
|
7 |
171 |
|
0.23 |
229 |
|
8 |
155 |
|
0.26 |
271 |
|
9 |
145 |
|
0.38 |
305 |
|
10 |
144 |
|
0.26 |
253 |
|
11 |
144 |
|
0.30 |
270 |
|
12 |
143 |
|
0.21 |
234 |
|
13q |
98 |
|
0.22 |
179 |
|
14q |
93 |
|
0.34 |
208 |
|
15q |
89 |
|
0.36 |
203 |
|
16 |
98 |
|
0.43 |
201 |
|
17 |
92 |
|
0.23 |
147 |
|
18 |
85 |
|
0.30 |
172 |
|
19 |
67 |
|
0.20 |
110 |
|
20 |
72 |
|
0.30 |
191 |
|
21q |
39 |
|
0.31 |
151 |
|
22q |
43 |
|
0.95 |
185 |
|
X |
164 |
|
0.31 |
231 |
|
Genome |
3154 |
|
0.31 |
208 |
|
|
|
|
|
|
|
Source: Adapted from McCarthy (1996).
Note: The data were obtained at 3000 rads. Hence physical distances relative to cR are roughly 8/3 as large as the data shown in Table 7.1.
a cM/cR scales the genetic and radiation hybrid maps. kb/cR scales the physical and radiation hybrid maps.
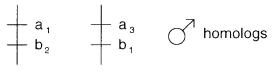
|
|
|
|
|
|
|
|
|
|
|
|
|
SINGLE-SPERM PCR |
217 |
||
order to analyze specific recombination events, it is necessary to be able to examine the |
|
|
|
|
||||||||||||
markers |
cosegregating in each single sperm cell. |
|
|
|
Sperm cannot be biologically amplified |
|
||||||||||
except by fertilization with an oocyte, and the impracticality of such experiments in the |
|
|
|
|
||||||||||||
human is all too apparent. Thus, until in vitro amplification became a reality, it was im- |
|
|
|
|
||||||||||||
possible to think of analyzing the DNA of single sperm. With PCR, however, this picture |
|
|
|
|
||||||||||||
has changed dramatically. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
A series of feasibility tests for single-sperm PCR was performed by Norman Arnheim |
|
|
|
|
||||||||||||
and co-workers. First they asked whether single diploid human cells could |
be |
genotyped |
|
|
|
|
|
|||||||||
by PCR. They mixed cells from donors who had normal hemoglobin, homozygous for the |
|
|
|
|
|
|
||||||||||
A |
gene with cells from donors who had sickle cell anemia, homozygous for the |
|
|
|
|
|
|
S |
gene. |
|||||||
Single cells were selected by micromanipulation and were tested by PCR amplification of |
|
|
|
|
|
|||||||||||
the |
locus followed by hybridization of a dot blot with allele-specific oligonucleotide |
|
|
|
||||||||||||
probes. The purpose of the experiment was to determine whether the pure alleles could be |
|
|
|
|
|
|
||||||||||
detected |
reliably, or whether there would |
be cross-contamination between |
the |
two types |
|
|
|
|
||||||||
of cells. The results for 37 cells analyzed were 19 |
|
|
|
A , 12 |
S , 6 none, and 0 both. This was |
|
||||||||||
very encouraging. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
The next test involved the analysis of single |
sperm |
from |
donors |
with two |
different |
|
|
|
|
|||||||
LDL receptor alleles. For 80 sperm analyzed, the results were 22 allele 1, 21 allele 2, 1 |
|
|
|
|||||||||||||
both alleles, and the remainder neither allele. Thus the efficiency of the single-sperm |
|
|
|
|||||||||||||
analysis |
was less than with diploid cells, but |
the cross-contamination |
level |
was low |
|
|
|
|||||||||
enough to be easily dealt with. A final test was to look at two nonlinked two-allele sys- |
|
|
|
|||||||||||||
tems. One was on chromosome 6 (a |
|
1 |
or a |
2 ), and |
the |
other |
on |
chromosome 19 |
(b |
1 |
or b 2 ). |
|||||
Four types of gametes were expected in equal ratios. What was actually observed is |
|
|
|
|
|
|
|
|||||||||
|
|
|
a 1b 1 |
|
a 1b 2 |
|
a 2 b 1 |
|
|
a 2 b 2 |
|
|
|
|||
|
|
|
21 |
18 |
|
14 |
|
|
|
17 |
|
|
|
|
||
This is really quite close to what was expected statistically, |
|
|
|
|
|
|
|
|
|
|
|
|||||
The final step was to do single-sperm linkage studies. In this case one does simultane- |
|
|
|
|||||||||||||
ous PCR analyses on linked markers. Consider the example shown in Figure 7.8. Nothing |
|
|
|
|
||||||||||||
needs to be known about the parental haplotypes to start with. PCR |
analysis |
automati- |
|
|
|
|
||||||||||
cally provides the parental phase. In the case shown in the figure, most of the sperm mea- |
|
|
|
|
||||||||||||
sured at loci a and b have either alleles a |
|
|
1 and b |
2 |
or a |
3 and b |
1. This indicates that these are |
|
||||||||
the haplotypes of the two homologous chromosomes in the donor. Occasional recombi- |
|
|
|
|
|
|||||||||||
nants |
are |
seen with alleles a |
1 and b |
1 or a |
3 and |
b |
2 . The frequency |
at which |
these |
are ob- |
|
|||||
served is a true measure of the male meiotic recombination frequency between markers a and b. Since large numbers of sperm can be measured, one can determine this frequency very accurately. Perhaps more important, with large numbers of sperm, very rare meiotic
recombination events can be detected, and thus very short genetic distances can be measured.
Figure 7.8 Two unrecombined homologous chromosomes that should predominate in the sperm expected from a hypothetical donor. Note that the observed pattern of alleles also determines the phase at these loci if it is not known in advance.