

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
8 Physical Mapping
WHY HIGH-RESOLUTION PHYSICAL MAPS ARE NEEDED |
|
Physical maps are needed because ordinary human genetic maps are |
not detailed enough |
to allow the DNA that corresponds to particular genes to be isolated efficiently. Physical |
|
maps are also needed as the source for the DNA samples that can serve as the actual sub- |
|
strate for large-scale DNA sequencing projects. Genetic linkage |
mapping provides a set |
of ordered markers. In experimental organisms this set can |
be almost as dense as one |
wishes. In humans one is much more limited because of the inability to control breeding |
|
and produce large numbers of offspring. Distances that emerge from human genetic map- |
|
ping efforts are vague because of the uneven distribution of meiotic recombination events |
|
across the genome. Cytogenetic mapping, until recently, was low resolution; it is improving now, and if it were simpler to automate, it could well provide a method that would supplant most others. Unfortunately, current conventional approaches to cytogenetic map-
ping seem difficult to automate, and they are slower than many other approaches that do not involve image analysis.
It is worth noting here that in some other organisms, cytogenetics is more powerful than in the human. For example, in Drosophila the presence of polytene salivary gland chromosomes provides a tool of remarkable power and simplicity. The greater extension
and higher-resolution imaging of polytene chromosomes allows bands to be seen at a typ-
ical resolution of 50 kb (Fig. 8.1). This is more than 20 times the resolution in the best human metaphase FISH. Furthermore the large number of DNA copies in the metaphase
salivary chromosomes means that microdissection and cloning are much more powerful
here than they are in the human. It would be nice if there were a convenient way to place large continuous segments of human DNA into Drosophila and then use the power of the cytogenetics in this simple organism to map the human DNA inserts.
Radiation hybrids offer, in principle, a way to measure distances between markers accurately. However, in practice, the relative distances in radiation hybrid maps appear to be distorted (Chapter 7). In addition there are a considerable number of unknown features of
these cells; for example, the |
types |
of unseen |
DNA rearrangements that may be present |
|||
need to be characterized. Thus it |
is not |
yet clear |
that the use of radiation hybrids |
alone |
||
can |
produce a complete |
accurate map or yield a set |
of DNA samples worth subcloning |
|
||
and |
characterizing at |
higher resolution. Instead, |
currently other methods must be used |
to |
||
accomplish the three major goals in physical mapping:
1.Provide an ordered set of all of the DNA of a chromosome (or genome).
2.Provide accurate distances between a dense set of DNA markers.
3.Provide a set of DNA samples from which direct DNA sequencing of the chromo-
some or genome is possible. This is sometimes called a |
sequence-ready map. |
234
RESTRICTION MAPS |
235 |
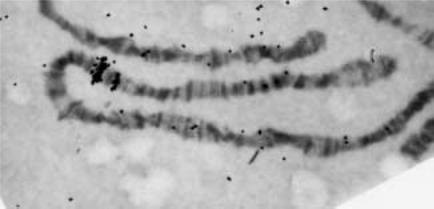
Figure 8.1 Banding seen in polytene Drosophila salivary gland chromosomes. Shown is part of chromosome 3 of for which a radiolabeled probe to the gene for heat-shock protein hsp70 has been hybridized. A strong and weak site are seen. (Figure kindly provided by Mary Lou Pardue.)
Most workers in the field focus their efforts on particular chromosomes or chromosome regions. Usually a variety of different methods are brought to bear on the problems
of isolating clones and probes and using these to order each other and ultimately produce finished maps. However, some research efforts have produced complete genomic maps with a restricted number of approaches. Such samples are turned over to communities interested in particular regions or chromosomes that finish a high-resolution map of the regions of interest.
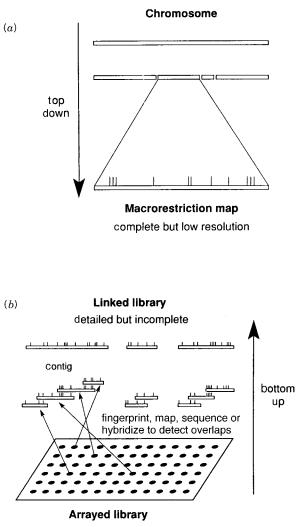
There are basically two kinds of physical maps commonly used in genome studies: restriction maps and ordered clone banks or ordered libraries. We will discuss the basic methodologies used in both approaches separately, and then, in Chapter 9, we will show how the two methods can be merged in more powerful second generation strategies. First
we outline the relative advantages and disadvantages of each type of map.
RESTRICTION MAPS
A typical restriction map is shown in Figure 8.2. It consists of an ordered set of DNA fragments that can be generated from a chromosome by cleavage with restriction enzymes individually or in pairs. Distances along the map are known as precisely as the lengths of the DNA fragments generated by the enzymes can be measured. In practice,
the |
lengths are measured |
by electrophoresis in virtually all currently used methods; |
they |
are accurate to a |
single base pair up to DNAs around around 1 kb in sizes. |
Lengths can be measured with better than 1% accuracy for fragments up to 10 kb, and
with a low percent of accuracy for fragments up to 1 Mb |
in size. Above this length, |
|
measurements today are still fairly qualitative, |
and it is |
always best to try to subdivide |
a target into pieces less than 1 Mb before any |
quantitative claims are made about its |
|
true total size. |
|
|

236 PHYSICAL MAPPING
Figure 8.2 Typical section of a restriction map generated by digestion of genomic or cloned DNA with two enzymes with different recognition sites A, and N.
In an ideal restriction map each DNA fragment is pinned |
to markers on other maps. |
||
Note that |
if this is |
done with randomly chosen probes, |
the locations of these probes |
within each |
DNA fragment |
are generally unknown (Fig. 8.3). |
Probes that correspond to |
the ends of the DNA fragments are more useful, when they are available, because their position on the restriction map is known precisely. Originally probes consisted of unsequenced DNA segments. However, the power of PCR has increasingly favored the use of sequenced DNA segments.
A major advantage of a restriction map is that accurate lengths are known between sets of reference points, even at very early stages in the construction of the map. A second advantage is that most restriction mapping can be carried out using a top-down strategy that
preserves an overview of the target and that reaches a nearly |
complete |
map relatively |
quickly. A third advantage of restriction mapping is that one |
is working |
with genomic |
DNA fragments rather than cloned DNA. Thus all of the potential artifacts that can arise |
||
from cloning procedures are avoided. Filling in the last few small pieces is always a chore |
||
in restriction mapping, but the overall map is a useful tool long before this is accomplished, and experience has shown that restriction maps can be accurately and completely constructed in reasonably short time periods.
In top-down mapping one successively divides a chromosome target into finer regions and orders these (Fig. 8.4). Usually a chromosome is selected by choosing a hybrid cell in which it is the only material of interest. There have been some concerns about the use of
hybrid cells as the source of DNA for mapping projects. In a typical hybrid cell there is no compelling reason for the structure of most of the human DNA to remain intact. The biological selection that is used to retain the human chromosome is actually applicable to
only a single gene on it. However, the available results, at least for chromosome 21, indicate that there are no significant differences in the order of DNA markers in a selected set
of hybrid and human cell lines (see Fig. 8.49). In a way this is not surprising; even in a human cell line most of the human genome is silent, and if loss or rearrangement of DNA
were facile under these circumstances, |
it should have been observed. Of course, for |
model organisms with small genomes, there is no need to resort to a hybrid cell at all— |
|
their genomes can be studied intact, or the chromosomes can be purified in bulk by PFG. |
|
In a typical restriction mapping effort, any preexisting genetic map information can be |
|
used as a framework for constructing the physical map. Alternatively, the chromosome of |
|
interest can be divided into regions by |
cytogenetic methods or low-resolution FISH. |
Figure 8.3 Ambiguity in the location of a hybridization probe on a DNA fragment.
RESTRICTION MAPS |
237 |
Figure 8.4 |
Schematic |
illustration of methods used in physical mapping. ( |
a ) Top-down strategy |
used in restriction mapping. ( |
b ) Bottom-up strategy used in making an ordered library. |
|
|
Large DNA fragments are produced by cutting the chromosome with restriction enzymes |
|
|
|
||||||||||
with very rare recognition sites. The fragments are separated by size and assigned to re- |
|
|
|||||||||||
gions by hybridization with genetically or cytogenetically mapped DNA probes. Then the |
|
|
|
||||||||||
fragments are assembled into contiguous blocks, by |
methods that will be described |
later |
|
|
|||||||||
in this chapter. The result at this point |
is |
called |
a |
|
|
macrorestriction map. |
The fragments |
||||||
may average 1 Mb in size. For a simple genome this means that only 25 fragments will |
|
|
|||||||||||
have to be ordered. This is relatively straightforward. For an intact human genome, the |
|
|
|||||||||||
corresponding number is 3600 fragments. This is an |
unthinkable |
task unless |
the frag- |
|
|||||||||
ments are first assorted into individual chromosomes. |
|
|
|
|
|
|
|
|
|||||
If a finer |
map |
is desired, it |
can |
be constructed by taking |
the |
ordered |
fragments |
one |
|
||||
at a time, |
and |
dissecting these |
with |
more |
frequently |
cutting |
restriction |
nucleases. |
An |
|
|||
238 |
|
|
PHYSICAL |
MAPPING |
|
|
|
|
|
|
advantage |
of |
this reductionist mapping approach is that the finer maps can be made only |
||||||||
in |
those regions where there is sufficient interest to justify this much more arduous task. |
|||||||||
|
The major disadvantage of most restriction mapping efforts is that they do not produce |
|||||||||
the |
DNA |
in |
a |
convenient, immortal |
form |
where |
it can be distributed or sequenced |
|||
by |
available methods. One could try to clone the large DNA fragments that compose |
|||||||||
the |
macrorestriction map, and there has been some progress in developing the vectors |
|||||||||
and |
techniques |
needed |
to do this. One could also use PCR |
to |
generate segments of |
|||||
these large fragments (see Chapter 14). For a small genome, most of the macrorestriction |
||||||||||
fragments it contains can usually be separated and purified by a single PFG fractionation. |
||||||||||
An |
example |
is |
shown |
in Figure 8.5. |
In |
cases |
like this, |
one |
does really possess the |
|
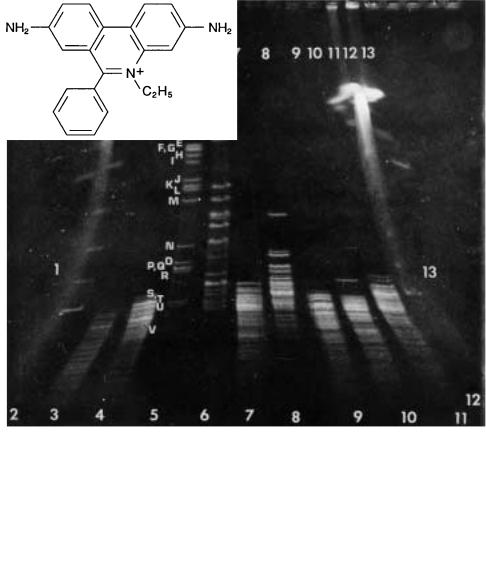
Figure 8.5 Example of the fractionation of a |
|
||||||||
restriction |
enzyme |
digest |
of |
an |
entire |
small |
|
||
genome |
by |
PFG. |
|
Above: |
Not |
|
I digest of |
the |
|
4.6 |
Mb |
E. coli |
genome |
shown |
in lane |
5; |
Sfi I |
||
digest |
in |
lane 6. |
Other |
lanes |
shown |
enzymes |
|
||
that cut too frequently to |
be useful for |
|
map- |
|
|||||
ping. (Adapted from Smith et al., 1987.) |
|
Left: |
|||||||
Structure of the ethidium cation used to stain the |
|
||||||||
DNA fragments. |
|
|
|
|
|
|
|
||

ORDERED LIBRARIES |
239 |
genome, but it is not in a form where it is easy to handle by most existing techniques. For a large genome, PFG does not have sufficient resolution to resolve individual macrore-
striction fragments. If one starts instead with a hybrid |
cell, containing only a single hu- |
||||
man chromosome or chromosome fragment, most of the |
human macrorestriction frag- |
||||
ments will be separable from one another. But they will |
still be contaminated, each by |
||||
many other background fragments from the rodent host. |
|
|
|||
ORDERED |
LIBRARIES |
|
|
|
|
Most genomic libraries are made by partial digestion with a relatively frequently cutting |
|||||
restriction |
enzymes, size selection of the fragments |
to provide a fairly uniform set of |
|||
DNA inserts, and then cloning these into a vector appropriate for the size range of inter- |
|||||
est. Because of the method by which |
they were produced, the cloned fragments are a |
||||
nearly random set of DNA pieces. Within the library, a given small DNA region will be |
|||||
present on many different clones. These extend to varying degrees on both sides of the |
|||||
particular region (Fig. 8.6). Because the clones |
contain overlapping regions of the |
||||
genome, it is possible to detect these overlaps by various fingerprinting methods that ex- |
|||||
amine patterns of sequence on particular clones. The random nature of the cloned frag- |
|||||
ments means that many more clones exist than the minimum set necessary to cover the |
|||||
genome. In practice, the redundancy of the library is usually set at fiveto tenfold in order |
|||||
to ensure that almost all regions of the genome will have been sampled at least once (as |
|||||
discussed in Chapter 2). From this vast library the goal is to assemble and to order the |
|||||
minimum set of clones that covers the genome in one contiguous block. This set is called |
|||||
the |
tiling path. |
|
|
|
|
|
Clone libraries have usually been ordered by a bottom-up approach. Here individual |
||||
clones are initially selected from the |
library at random. Usually the library is handled as |
||||
an array of samples so that each clone has a unique location on a set of microtitre plates, |
|||||
and the chances of accidentally confusing two different |
clones can be minimized. The |
||||
clone is fingerprinted, by hybridization, by restriction mapping, or by determining bits of |
|||||
DNA sequence. Eventually clones appear that share some or all of the same fingerprint |
|||||
pattern (Fig. 8.7). These are clearly overlapping, if not identical, and they are assembled |
|||||
into |
overlapping sets, called |
contigs, |
which is short for contiguous blocks. There are sev- |
||
eral obvious advantages to this approach. Since the DNA is handled as clones, the map is |
|||||
built up of immortal samples that are |
easily distributed |
and that are potentially suitable |
|||
for direct sequencing. The maps are usually fairly high resolution when small clones are |
|||||
used, and some forms of fingerprinting provide very |
useful internal information about |
||||
each clone. |
|
|
|
|
|
Figure 8.6 |
Example of a dense library of clones. ( |
a ) The large number of clones insures that a |
||
given DNA probe or region (vertical |
dashed line) will occur on quite a few different clones in the li- |
|||
brary. ( |
b ) The minimum tiling |
set is the smallest number of clones that can be selected to span the |
||
entire sample of DNA. |
|
|
|
|
240 PHYSICAL MAPPING
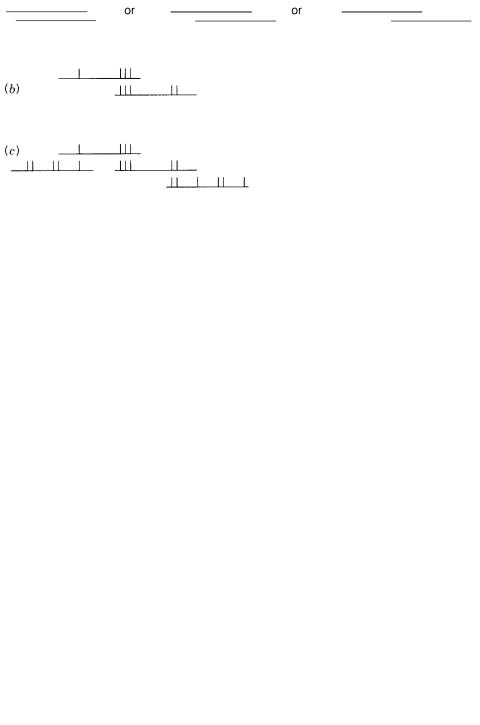
Figure 8.7 |
Example of a bottom-up |
fingerprinting strategy |
to order |
a dense set of clones. ( |
a ) A |
clone is selected at random and fingerprinted. ( |
|
b ) |
Two clones that share an overlapping fingerprint |
||
pattern are assembled into a contig. ( |
c ) Longer |
contigs |
are assembled as more overlapping |
clones |
|
are found. |
|
|
|
|
|
There |
are a |
number |
of disadvantages to bottom-up |
mapping. |
While |
this |
process |
is |
||||
easy to automate, no overview of the |
chromosome |
or genome is |
|
provided |
by |
the |
||||||
fingerprinting |
and |
contig |
building. Additional experiments have to |
be |
done |
to |
place |
|||||
contigs on |
a |
lower-resolution framework map, and most approaches do |
not necessarily |
|||||||||
allow the |
orientation of |
each contig to be |
determined |
easily. A |
more |
serious |
limitation |
|||||
of pure bottom-up mapping strategies is that they do not reach completion. Even if the original library is a perfectly even representation of the genome, there is a statistical prob-
lem |
associated with the random clone picking used. After a while, most new clones |
that |
are |
picked will fall into contigs that are already saturated. No new information |
will be |
gained from the characterization of these new clones. As the map proceeds, a diminishingly smaller fraction of new clones will add any additional useful information. This problem becomes much more serious if the original library is an uneven sample of the chromosome or genome. The problem can be alleviated somewhat if mapped contigs are
used to screen new clones prior to selection to try to discard those that cannot yield new information. An additional final problem with bottom-up maps is shown in Figure 8.8.
Usually the overlap distance between two |
adjacent members of |
a |
contig |
is not known |
|
with much precision. Therefore distances |
on a typical bottom-up |
map are |
not |
well de- |
|
fined. |
|
|
|
|
|
The number of samples or clones that must be handled in top-down or bottom-up mapping |
|||||
projects can be daunting. This number also |
scales linearly with |
the |
resolution |
desired. To |
|
gain some perspective on the problem, consider the task of mapping a 150-Mb chromosome.
Figure 8.8 Ambiguity in the degree of clone overlap resulting from most fingerprinting or clone ordering methods.
RESTRICTION NUCLEASE GENOMIC DIGESTS |
241 |
This is the average size of |
a human chromosome. The |
numbers of samples needed are |
|
|
|
shown below: |
|
|
|
|
|
RESOLUTION |
RESTRICTION MAP |
|
ORDERED LIBRARY |
(5 REDUNDANCY |
) |
1 Mb |
150 fragments |
|
750 clones |
|
|
0.1 Mb |
1500 fragments |
|
7500 clones |
|
|
0.01 Mb |
15,000 fragments |
75,000 clones |
|
||
With existing methods the current convenient range for constructing physical maps of en- |
|
|
|||
tire human chromosomes allows a resolution of somewhere between 50 kb (for the most |
|
|
|||
arduous bottom-up approaches attempted) to 1 Mb for much easier restriction mapping or |
|
|
|||
large insert clone contig building. |
|
|
|
|
|
The resolution desired in a map will determine the sorts of clones that are conveniently |
|
|
|||
used in bottom-up approaches. Among the possibilities currently available are |
|
|
|||
Bacteriophage lambda |
|
10 kb inserts |
|
|
|
Cosmids |
|
|
40 kb inserts |
|
|
P1 clones |
|
|
80 to 100 kb inserts |
|
|
Bacterial artificial chromosomes (BACs) |
100 to 400 kb inserts |
|
|
||
Yeast artificial chromosomes (YACs) |
|
100 to 1300 kb inserts |
|
|
|
The first three types of clones can be easily grown in |
|
large numbers of copies per cell. |
|
|
|
This greatly simplifies DNA preparation, hybridization, and other analytical procedures. |
|
|
|||
The last two types of clones are usually handled as single copies per host cell, although |
|
|
|||
some methods exist for amplifying them. Thus they are more difficult to work with, indi- |
|
|
|||
vidually, but their larger insert size makes low-resolution mapping much more rapid and |
|
|
|||
efficient. |
|
|
|
|
|
RESTRICTION NUCLEASE GENOMIC |
DIGESTS |
|
|
|
|
Generating DNA fragments of the desired size range is critical for producing useful li- |
|
|
|||
braries and genomic restriction maps. If genomes were statistically random collections of |
|
|
|||
the four bases, simple binomial statistics would allow us to estimate the average fragment |
|
|
|||
length that would result from a given restriction nuclease recognition site in a total digest. |
|
|
|||
For a genome where each of the four bases is equally represented, the probability of oc- |
|
|
|||
currence of a particular site of size |
n |
is 4 n ; therefore the average fragment length gener- |
|
||
ated by that enzyme will be 4 |
n . In practice, this becomes |
|
|
||
|
SITE SIZE (N ) |
|
AVERAGE FRAGMENT LENGTH |
(kb) |
|
|
4 |
|
1 |
|
|
|
6 |
|
4 |
|
|
|
8 |
|
64 |
|
|
|
10 |
|
1000 |
|
|
242 |
|
PHYSICAL |
MAPPING |
|
|
|
|
|
|
|
|
|
|
|
|
|
This tabulation indicates that enzymes with four or six base sites are convenient for the |
|
|
|
|||||||||||||
construction of partial digest small insert libraries. Enzymes with sites |
ten bases |
long |
|
|
|
|||||||||||
would be the preferred choice for low-resolution macrorestriction mapping, but such en- |
|
|
|
|
||||||||||||
zymes are unknown. Enzymes with eight-base sites would be most useful for currently |
|
|
|
|
||||||||||||
achievable large-insert cloning. More accurate schemes for predicting cutting frequencies |
|
|
|
|
||||||||||||
are discussed in Box 8.1. |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
A list of the enzymes currently available that have relatively rare cutting sites in mam- |
|
|
|
||||||||||||
malian genomes is given in Table 8.1. Unfortunately, there are only a few known restriction |
|
|
|
|
||||||||||||
enzymes with eight-base specificity, and most of these have sites that are not well-predicted |
|
|
|
|
||||||||||||
by random statistics. A few enzymes are known with larger recognition sequences. In most |
|
|
|
|
|
|||||||||||
cases there is not yet convincing evidence that the available |
preparations of |
these |
enzymes |
|
|
|
|
|||||||||
have low enough contamination with random nonspecific nucleases to allow them to be used |
|
|
|
|
|
|
||||||||||
for a complete digest to generate a discrete nonoverlapping |
set of large DNA fragments. |
|
|
|
|
|||||||||||
Several enzymes exist that have sites so rare that they |
will not occur at all |
in |
natural |
|
|
|||||||||||
genomes. To use these enzymes, one must employ strategies in which the sites are introduced |
|
|
|
|
|
|||||||||||
into the genome, their location is determined, and then cutting at the site is used to generate |
|
|
|
|||||||||||||
fragments containing segments of the region where the site was inserted. Such strategies are |
|
|
|
|
||||||||||||
potentially quite powerful, but they are still in their infancy. |
|
|
|
|
|
|
|
|
||||||||
|
The unfortunate conclusion, from experimental studies on the pattern of DNA fragment |
|
|
|
|
|||||||||||
lengths generated by genomic restriction nuclease digestion, |
is that most currently available |
|
|
|
|
|||||||||||
8-base specific enzymes are not useful for generating fragments suitable for macrorestriction |
|
|
|
|
|
|||||||||||
mapping. Either the average fragment length is too short, or the digestion is not complete, |
|
|
|
|||||||||||||
leading to an overly complicated set of reaction products. However, mammalian genomes are |
|
|
|
|
|
|||||||||||
very poorly modeled by binomial statistics, |
and |
thus |
some |
enzymes, |
which |
statistically |
|
|
|
|||||||
might be thought to be useless because they would generate fragments that are too small, in |
|
|
|
|
||||||||||||
fact generate fragments in useful size ranges. As a specific example, consider the first two en- |
|
|
|
|
||||||||||||
zymes known with eight-base recognition sequences: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
Sfi |
I |
|
|
GGCCN^NNNNGGCC |
|
|
|
|
|
|
||
|
|
|
|
Not |
I |
|
|
GC^GGCCGC |
|
|
|
|
|
|
|
|
Here the symbol N indicates that any of the four bases can occupy this site; the caret (^) |
|
|
|
|||||||||||||
indicates the cleavage site on the strand shown; there is |
a |
corresponding site |
on the sec- |
|
|
|
||||||||||
ond strand. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Human |
DNA |
is A–T |
rich; like that of other |
mammals |
it |
contains approximately |
60% |
|
|
|
|||||
A T. When this is factored into the predictions (Box 8.1), on the basis of base composi- |
|
|
||||||||||||||
tion, alone, these enzymes would be expected to cut human DNA every 100 to 300 kb. In |
|
|
||||||||||||||
fact |
Sfi |
I digestion of the human genome yields DNA fragments that predominantly range |
|
|
|
|
||||||||||
in size from 100 to 300 kb. In contrast, |
|
|
Not |
I generates |
DNA |
fragments that average |
|
|||||||||
closer to 1 Mb in size. The size range generated by |
|
|
|
|
Sfi |
I would make it potentially useful |
||||||||||
for some applications, but the cleavage specificity of |
|
|
|
|
Sfi |
I leads to some unfortunate com- |
||||||||||
plications. This enzyme cuts within an unspecified sequence. Thus the fragments it gener- |
|
|
|
|
||||||||||||
ates cannot be directly cloned in a straightforward manner |
because the three base over- |
|
|
|
|
|||||||||||
hang |
generated |
by |
Sfi I is |
a mixture |
of 64 different sequences. Another problem, |
|
||||||||||
introduced by the location of the |
|
Sfi |
I cutting site, is |
that different |
sequences |
turn out |
to |
|||||||||
be cut at very different rates. This makes it difficult to achieve total digests efficiently, and |
|
|
|
|||||||||||||
as described later, it also makes it very difficult to use |
|
|
|
Sfi |
I in |
mapping |
strategies |
that de- |
||||||||
pend on the analysis of partial digests. |
|
|
|
|
|
|
|
|
|
|
|
|
||||

RESTRICTION NUCLEASE GENOMIC DIGESTS |
243 |
BOX 8.1
PREDICTION OF RESTRICTION ENZYME-CUTTING FREQUENCIES
An accurate prediction of cutting |
frequencies requires an accurate statistical estima- |
||||
tion of the probability of occurrence of the enzyme recognition site. To accomplish |
|||||
this, one must take into account two factors: First, the sample of interest, that is, the |
|||||
human genome, is unlikely to have a base composition that is precisely 50% G |
C, |
||||
50% A T. Second, |
the |
frequencies of particular dinucleotide sequences often vary |
|||
quite substantially from that predicted by simple binomial statistics based on the base |
|||||
composition. A rigorous treatment should also take the mosaicism into account (see |
|
||||
Chapters 2 and 15). |
|
|
|
|
|
Base composition effects alone can be considered, for double-stranded DNA with a |
|||||
single variable: |
X G |
C |
1 X A T , where X is a mole fraction. Then the expected fre- |
||
quency of occurrence of a site with |
n G’s or C’s and |
m |
A’s or T’s is just |
||
|
2 |
|
|
|
|
|
|
|
|
1 |
n m (X G |
C )n (1 X G C )m |
|
||||
|
|
|
||||||
To take base sequence into account, Markov statistics can be used. In Markov chain |
|
|||||||
statistics the probability of the |
|
n th event in a |
series can |
be |
influenced by the |
specific |
||
outcome of the prior events such as the |
( |
|
n |
1)th |
and |
( |
n 2)th |
events. Thus Markov |
chain statistics can take into account known frequency information about the occur- |
|
|
||||||
rences of sequences of events. In other |
words, this kind |
of statistics |
is ideally |
suited |
|
|||
for the analysis of sequences. |
|
|
|
|
|
|
|
|
Suppose that the frequencies of particular dinucleotide sequences are known for the |
|
|
||||||
sample of interest. There are 16 possible dinucleotide sequences: The frequency |
|
X A,C, |
||||||
for example, indicates the fraction |
of dinucleotides that has a AC sequence on |
one |
|
|||||
strand base paired to a complementary GT on the other. The sum of these frequencies |
|
|
||||||
is 1. Only 10 of the 16 dinucleotide sequences are distinct unless we consider the two |
|
|||||||
strands separately. On each strand, we can relate dinucleotide and mononucleotide fre- |
|
|||||||
quencies by four equations: |
|
|
|
|
|
|
|
|
|
X A,C X A,A |
X A,T X A,G X A |
|
|||||
since base A must always be followed by some other base (the X’s indicate mole fraction). The expected frequency of occurrence of a particular sequence string, based on these nearest-neighbor frequencies is just
|
n |
X i,i 1 |
|
|
X 12 |
|
|
|
X i |
||
|
i 2 |
||
where the product is taken successively over the mole fractions |
|
|
|
X i of all successive dinucleotides |
i,i 1, and mononucleotides |
||
terest. Predictions done in this way are usually more accurate than predictions based
solely on the base composition. Where |
methylation occurs that can block the cutting |
|
of |
certain restriction nucleases, the |
issue becomes much more complex, as discussed |
in |
the text. |
|
X i,i 1 , respectively, and i in the sequence of in-