

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
1 DNA Chemistry and Biology
BASIC PROPERTIES OF DNA
DNA is one of the fundamental molecules of all life as we know it. Yet many of the features of DNA as described in more elementary sources are incorrect or at least misleading. Here a
brief but fairly rigorous overview will be presented. Special stress will be given to try to clarify any misconceptions the reader has based on prior introductions to this material.
COVALENT STRUCTURE
The basic chemical structure of DNA is well-established. It is shown in Figure 1.1. Because the phosphate-sugar backbone of DNA has a polarity, at each point along a polynucleotide
chain the direction of that |
chain is always uniquely defined. It proceeds from the 5 |
|
-end via |
|||
3 - to |
5 -phosphodiester |
bonds until the 3 |
-end is reached. The |
structure of DNA shown in |
||
Figure 1.1 is too elaborate to make this representation useful for larger segments of DNA. |
||||||
Instead, we abbreviate this structure by a series of shorthand forms, as shown in Figure 1.2. |
||||||
Because of the polarity of the DNA, it is important to realize that different sequences of |
||||||
bases, in our abbreviations, actually correspond to different chemical structures (not simply |
||||||
isomers). So ApT and TpA are different compounds with, occasionally, rather different prop- |
||||||
erties. The simplest way to abbreviate DNA is |
to draw a single polynucleotide strand |
as |
a |
|||
line. Except where explicitly stated, this |
is always drawn so that the left-hand |
end corre- |
||||
sponds to the 5 |
-end of the molecule. |
|
|
|
||
RNA differs from DNA by having an additional hydroxyl at the 2 |
|
-position of the sugar |
||||
(Fig. 1.1). This has two major implications that distinguish the chemical and physical proper- |
||||||
ties of RNA and DNA. The 2 |
|
-OH makes RNA unstable with respect to alkaline hydrolysis. |
||||
Thus RNA is a molecule intrinsically designed for turnover at the slightly alkaline pH’s nor- |
||||||
mally found in cells, while DNA is chemically far more stable. The 2 |
|
-OH also restricts the |
||||
range of energetically favorable conformations of the sugar ring and the phosphodiester back- |
|
|||||
bone. This limits the range of conformations of the RNA chain, compared to DNA, and it ul- |
||||||
timately restricts RNA to a much narrower choice of helical structures. Finally the 2 |
|
-OH can |
||||
participate in interactions with phosphates or bases that stabilize folded chain structures. As a |
||||||
result an RNA can usually attain stable tertiary structures (ordered, three-dimensional, rela- |
||||||
tively compact structures) with far more ease than the same corresponding DNA sequence. |
|
|
||||
DOUBLE |
HELICAL |
STRUCTURE |
|
|
|
|
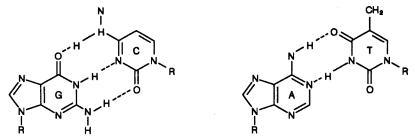
The two common base pairs A–T and G–C are well-known, and little evidence for other base interactions within the DNA’s double helix exists. The key feature of double-helical DNAs (duplexes), which dominates their properties, is that an axis of symmetry relates the two strands (Box 1.1).
1

2 DNA CHEMISTRY AND BIOLOGY
Figure 1.1 |
Structure of the phosphodiester backbone of DNA and RNA, and the four major bases |
|
||
found in DNA and RNA: the purines, adenine (A) and guanine (G), and the pyrimidines, cytosine (C) |
|
|||
and thymine (T) or uracil (U). |
(a) In these abbreviated structural formulas, every vertex not occupied by |
|
||
a letter is a carbon atom. R is H in DNA, OH in RNA; B and B |
indicate one of the four bases. |
(b) The |
||
vertical arrows show the base atoms that are bonded to the C1 |
carbon atoms of the sugars. |
|
||
In ordinary double-helical DNA this is a pseudo C2 axis, since it applies only to the |
|
|
|||||
backbones and not to the bases themselves, as shown in Figure 1.3 |
|
a. |
However, certain DNA |
|
|||
sequences, called |
self-complementary, |
have |
a true C2 symmetry axis perpendicular to |
|
|||
the helix axis. We frequently abbreviate DNA duplexes as pairs of |
parallel lines, |
as shown |
|
|
|||
in Figure 1.3 |
b. |
By convention, the |
top line almost |
always runs from |
5 |
-end to 3 |
-end. |
Figure 1.2 |
Three ways in which the structures of DNA and RNA are abbreviated. Note that BpB |
|
|
is not the same chemical compound as B |
pB. |
|
|
DOUBLE HELICAL STRUCTURE |
3 |
|
BOX 1.1 |
|
|
|
|
|
|
|
||
|
C2 |
SYMMETRY |
|
|
|
|
|
|
|
|
|
C2 symmetry implies that a structure is composed of two identical parts. An axis of rota- |
|
|
|||||||
|
tion can be found that interchanges these two parts by a 180-degree rotation. This axis is |
|
|
|||||||
|
called a |
C2 axis. |
An example of a common object that has such symmetry is the stitching |
|
|
|||||
|
of a baseball, which is used to bring two figure-8 shaped structures together to make a |
|
|
|||||||
|
spherical shell. An example of a well-known biological structure with C2 symmetry is the |
|
|
|||||||
|
hemoglobin tetramer which is made up of two identical dimers each consisting of one al- |
|
|
|||||||
|
pha chain and one beta chain. Another example is the streptavidin tetramer which con- |
|
|
|||||||
|
tains four copies of the same subunit and has three different C2 axes. One of these passes |
|
|
|||||||
|
horizontally right through the center of the structure (shown in Fig. 3.26). |
|
|
|
||||||
|
Helical symmetry means that a rotation and a translation along the axis of rotation |
|
|
|||||||
|
occur simultaneously. If that rotation is 180 degrees, the structure generated is called a |
|
|
|||||||
|
twofold |
helix |
or |
a |
pleated sheet. |
Such sheets, called |
beta structures, |
are commonly |
|
|
|
seen in protein structure but not in nucleic acids. The rotation that generates helical |
|
|
|||||||
|
symmetry |
does not |
need |
to be an integral fraction |
of 360 degrees; DNA |
structures |
|
|
||
|
have 10 to 12 bases per turn; under usual physiological conditions DNA shows an av- |
|
|
|||||||
|
erage |
of |
about 10.5 bases per turn. In the DNA double helix, the two strands wrap |
|
|
|||||
|
around a central helical axis at each turn. |
|
|
|
|
|||||
|
Pseudo C2 symmetry means that some aspects of a structure can be interchanged by a |
|
|
|||||||
|
rotation of 180 degrees, while other aspects of the structure are altered by this rotation. |
|
|
|||||||
|
This process might be imagined as a disk painted with the familiar yin and yang symbols |
|
|
|||||||
|
of the Korean flag. Then, except for a color change, a C2 axis perpendicular to the yin and |
|
|
|||||||
|
yang exchanges them. The pseudo C2 axes in DNA are perpendicular to the helix axis. |
|
|
|||||||
|
They occur in the plane of each base pair (this axis interchanges the position of the two |
|
|
|||||||
|
paired bases) and between each set of adjacent base pairs (this axis interchanges a base on |
|
|
|||||||
|
one strand with the nearest neighbor of the base to which it is paired to the other strand). |
|
|
|||||||
|
Thus for DNA with 10 bases per turn there are 20 C2 axes per turn. |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
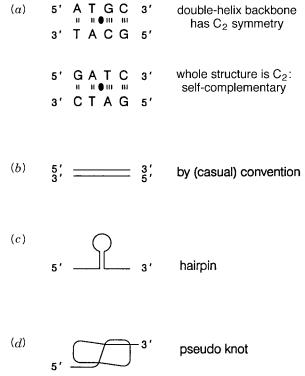
The antiparallel nature of the DNA strands imposed by the pseudosymmetry of their struc- |
|
|
ture means that the bottom strand in this simple representation runs in the opposite direction |
|
|
to our usual convention. Where the DNA sequence permits it, double helices can also be |
|
|
formed by the folding back of a single strand upon itself to make structures called |
|
hairpins |
(Fig. 1.3 c ) or more complex topologies such as structures called |
pseudoknots |
(Fig. 1.3 d ). |
4 DNA CHEMISTRY AND BIOLOGY
Figure 1.3 |
Symmetry and pseudosymmetry in DNA double helices. |
(a) The vertical lines indicate |
|
hydrogen bonds between base pairs. The base pairs and their attached backbone residues can be |
|||
flipped by a 180° rotation around an axis through the plane of the bases and perpendicular to the he- |
|||
lix axis (shown as a filled lens-shaped |
object). |
(b) Conventional way of representing a double- |
|
stranded DNA. |
(c) Example of a |
DNA hairpin. |
(d) Example of a DNA pseudoknot. |
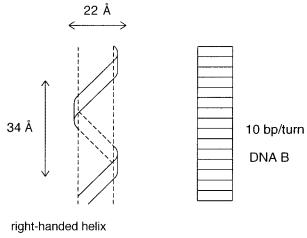
In a typical DNA duplex, the phosphodiester backbones are on the outside of the struc- |
|
|
ture; the base pairs are internal (Fig. 1.4). The structure of the double helix appears to be reg- |
|
|
ular because the A–T base pair fills a three-dimensional space in a manner similar to a G–C |
|
|
base pair. The spaces between the two backbones are called |
grooves. |
Usually one groove is |
much broader (the major groove) than the other (the minor groove). The structure appears as |
|
|
a pair of wires held fairly close together and wrapped loosely around a cylinder. Three major |
|
|
classes of DNA helical structures (secondary structures) have been found thus far. DNA B, |
|
|
the structure first analyzed by Watson and Crick, has 10 base pairs per turn. DNA A, which is |
|
|
very similar to the structure almost always present in RNA, |
has 11 base pairs per turn. Z |
|
DNA has 12 base pairs per turn; unlike DNA A and B, it is a left-handed helix. Only a very |
|
|
restricted set of DNA sequences appears able to adopt the Z helical structure. The biological |
|
|
significance of the range of structures accessible to particular DNA sequences is still not well |
|
|
understood. Elementary texts often focus on hydrogen bonding between the bases as a major |
|
|
force behind the stability of the double helix. The pattern of hydrogen bonds in fact is re- |
|
|
sponsible for the specificity of base–base interactions but |
not their stability. Stability is |
|
largely determined by electrostatic and hydrophobic interactions |
between parallel overlap- |
|
ping base planes which generate an attractive force called |
base stacking. |
|
METHYLATED BASES |
5 |
Figure 1.4 |
Schematic view of the three-dimensional structure of the DNA double helix. Ten base |
|
|
pairs per turn fill a central core; the two backbone chains are wrapped around this in a right-handed |
|
|
|
screw sense. Note that the A–T, T–A, G–C, and C–G base pairs all fit into |
exactly |
the same space |
|
between the backbones. |
|
|
|
METHYLATED |
BASES |
|
|
|
||
DNA from most higher organisms and from many lower organisms have additional ana- |
|
|
||||
logues of the normal bases. In bacteria these are principally |
|
N6 -methyl A and 5-methyl C |
||||
(or 4-methyl C). Higher organisms contain 5-methyl C. The presence of methylated bases |
||||||
has |
a |
strong |
biological effect. Once in place, the methylated bases are apt |
to |
maintain |
|
their |
methylated status after DNA replication. This is because the hemi-methylated du- |
|||||
plex |
produced |
by one round of DNA synthesis is a far |
better substrate for the enzymes |
|||
that insert the methyl groups (methylases) than the unmethylated sequence. The result is a |
||||||
form |
of inheritance (epigenetic) that goes beyond the |
ordinary DNA sequence: DNA |
has |
|||
a memory of where it has been methylated. |
|
|
|
|||
|
The role |
of these modified bases is understood in |
some detail. In bacteria these |
bases |
||
mostly arise by a postreplication endogenous methylation reaction. The purpose of methylation is to protect the cellular DNA against endogenous nucleases, directed against the same specific DNA sequences as the endogenous methylases. It constitutes a cellular defense or restriction system that allows a bacterial cell to distinguish between its own
DNA, |
which is methylated, |
and the DNA from an invading bacterial virus (bacterio- |
phage) |
or plasmid, which is |
unmethylated and can be selectively destroyed. Unless the |
host DNA is replicating too fast for methylation to keep up, it is continually protected by methylation shortly after it is synthesized. However, once lytic bacteriophages are successfully inside a bacterial cell, their DNA will also become methylated and protected
from |
destruction. In |
bacteria |
particular methylated sequences function to control initia- |
||||||
tion of DNA replication, DNA repair, gene expression, and movement of transposable el- |
|
||||||||
ements. |
|
|
|
|
|
|
|
|
|
|
In |
bacteria, |
methylases |
and |
their |
cognate nucleases |
recognize specific |
sequences |
that |
range |
in |
size from |
4 to 8 |
base |
pairs |
in length. Each |
site is independently |
methylated. |
In |
higher organisms the principal, if not the exclusive, site of methylation is the sequence CpG
6 |
DNA |
CHEMISTRY AND |
BIOLOGY |
|
|
|
|
|
|
|
|
which is |
converted to |
m CpG. Although the |
eukaryotic methylases recognize only |
a dinu- |
|
|
|
||||
cleotide sequence, methylation (or unmethylated) at CpGs appears to be regionally specific, |
|
|
|
|
|||||||
suggesting that nearby |
m CpG sequences interact. This plays a role in allowing cells with the |
|
|
||||||||
same exact DNA sequence to maintain stable, different patterns of gene expression (cell dif- |
|
|
|
|
|||||||
ferentiation), and it also allows the contributions of the genomes of two parents to be distin- |
|
|
|
|
|||||||
guished in offspring, since they often have different patterns of DNA methylation. |
|
|
|
|
|||||||
The fifth base in the DNA of humans and other vertebrates is 5-methyl C. Its presence |
|
|
|
|
|||||||
has profound consequences for the properties of these DNA. The influence of |
|
|
m |
C on DNA |
|||||||
three-dimensional structure is not yet fully explored. We know, however, that it favors the |
|
|
|
|
|||||||
formation of some unusual DNA structures, like the left-handed Z helix. Its biological |
|
|
|
|
|||||||
importance remains to be elucidated. However, it is on the level of the DNA sequence, the |
|
|
|
|
|||||||
primary structure, where the effect |
of |
|
m C is most profoundly felt. To |
understand |
the |
im- |
|
||||
pact, it is useful to consider why DNA contains the |
base T (5-methyl U) instead of U, |
|
|
|
|||||||
which predominates, overwhelmingly, in RNA. While the base T conveys a bit of extra |
|
|
|
|
|||||||
stability in DNA duplexes because of interactions between the methyl group and nearby |
|
|
|
|
|||||||
bases, the most decisive influence of the methyl group of T is felt in the repair of certain |
|
|
|
||||||||
potential mutagenic DNA lesions. By |
far the most common mutagenic DNA damage |
|
|
|
|
||||||
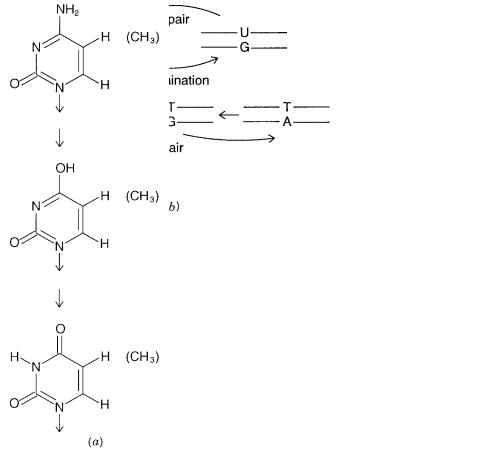
event in nature appears to be deamination of C. As shown in Figure 1.5, this yields, in du- |
|
|
|
||||||||
plex DNA, a U mispaired with a G. Random repair of this lesion would result, 50% of the |
|
|
|
||||||||
time, in replacement of the G by an A, a mutation, instead of replacement of the U by a |
|
|
|
||||||||
C, restoring the original sequence. |
|
|
|
|
|
|
|
||||
Apparently the intrinsic rate of the C to U mutagenic process is far too great for opti- |
|
|
|
||||||||
mum evolution. Some rate of mutation is always needed; otherwise, a species could not |
|
|
|
|
|||||||
adapt or evolve. Too high a rate can lead to deleterious mutations that interfere with re- |
|
|
|
|
|||||||
production. Thus the mutation rate must be carefully tuned. Nature accomplishes this for |
|
|
|
|
|||||||
deamination of C by a special repair system that recognizes the G–U mismatch and selec- |
|
|
|
|
|||||||
tively excises the U and replaces |
it with a C. This system, centered about an enzyme |
|
|
|
|||||||
called |
Uracil DNA glycosylase, |
biases the repair process in a way that effectively avoids |
|
||||||||
most mutations. However, a problem |
arises when the base 5- |
|
m C is |
present in the |
DNA. |
||||||
Then, as shown in Figure 1.5, |
deamination produces T which is a normally occurring |
|
|
|
|
||||||
base. Although the G–T mismatch is still repaired with a bias toward restoring the pre- |
|
|
|
|
|||||||
sumptive original |
m C, the process is not nearly as efficient (nor should it be, since some of |
|
|
||||||||
the time the G–T mismatch will have come by misincorporation of a G for an A). The re- |
|
|
|
|
|||||||
sult is that |
m C represents a mutation hotspot within DNA sequences that contain it. |
|
|
|
|
||||||
In the DNA of vertebrates, all known |
|
m C occur in the sequence |
|
m |
CpG. About 80% of |
||||||
this sequence occurs in the methylated form (on both strands). Strikingly the total occur- |
|
|
|
|
|||||||
rence of CpG (methylated or not) is only 20% of that expected from simple binomial sta- |
|
|
|
|
|||||||
tistics based on the frequency of occurrence of the four bases in DNA: |
|
|
|
|
|||||||
|
|
|
|
|
|
X CpG |
0.2 |
|
|
|
|
|
|
|
|
|
|
X C X G |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
where |
X indicates the mole fraction. The remainder of the expected CpG has apparently been |
|
|
|
|
||||||
lost through mutation. The occurrence of the product of the mutation, TpG, is elevated, as ex- |
|
|
|
|
|||||||
pected. This is a remarkable example of how a small bias, over the evolutionary time scale, |
|
|
|
|
|||||||
can lead to dramatic alteration in properties. Presumably the rate of mutation of |
|
|
m |
CpG’s con- |
|||||||
tinues to slow as the target size decreases. There must also be considerable functional con- |
|
|
|
|
|||||||
straints on the remaining |
|
m CpG’s that prevent their further loss. |
|
|
|
|
|||||
PLASTICITY IN DNA STRUCTURE |
7 |
Figure 1.5 |
Mutagenesis and repair processes that alter DNA sequences. |
(a) Deamination of C and |
||
m C |
produce |
U and T, respectively. |
(b) Repair of uracil-containing |
DNA can occur without errors, |
while repair of mismatched T–G pairs incurs some risk of mutagenesis. |
(c) Consequences of exten- |
|||
sive |
m CpG mutagenesis in mammalian DNA. |
|
|
|
The vertebrate immune system appears to have learned about the striking statistical ab- |
|
|
normality of |
vertebrates. Injections of DNA from other sources with a high G |
C con- |
tent, presumably with a normal ratio to CpG, act as an adjuvant; that is, these injections |
|
|
stimulate a generally heightened immune response. |
|
|
PLASTICITY |
IN DNA STRUCTURE |
|
Elementary discussions of DNA dwell on the beauty and regularity of the Watson-Crick double helix. However, the helical structure of DNA is really much more complex than this. The Watson-Crick structure has 10 base pairs per turn. DNA in solution under physi-
ological conditions shows an average structure of about 10.5 base pairs per turn, roughly halfway between the canonical Watson-Crick form and the A-type helix with a larger di-
ameter and tilted base pairs which are characteristic of RNA. In practice, these are just
8 |
DNA CHEMISTRY AND BIOLOGY |
|
|
|
|
average forms. DNA is revealed to be fairly irregular by methods that do not have to aver- |
|
||||
age over long expanses of structure. DNA is a very plastic molecule with a backbone eas- |
|
||||
ily distorted and with optimal geometry very much influenced by its local sequences. For |
|
||||
example, certain DNA sequences, like properly spaced sets of ApA’s promote extensive |
|
||||
curvature of the backbone. Thus, while base pairs |
predominate, the angle between the |
|
|||
base pairs, the extent of their stacking (which holds DNA together) above and below |
|
||||
neighbors, their planarity, and their disposition relative to helix axis can vary substan- |
|
||||
tially. Almost all known DNA structures can be viewed in detail by accessing the Nucleic |
|
||||
Acid Database, NDB |
http://ndbserver.rutgers.edu/ |
. |
|
||
|
We do not really know |
enough about the properties of proteins that |
recognize DNA. |
|
|
One extreme view is that these proteins look at the bases directly and, if necessary, distort |
|
||||
the helix into a form that fits well with the structure of protein residues in contact with the |
|
||||
DNA. The other extreme view has a key role played by the DNA structure with proteins |
|
||||
able to recognize structural variants, without explicit consideration of the sequence that |
|
||||
generated them. These views have very different implications for proteins that might rec- |
|
||||
ognize classes of DNA sequences rather than just |
distinct single sequences. We are not |
|
|||
yet able to decide among these alternative views or to adopt some sort of compromise po- |
|
||||
sition. The structures of the few protein-nucleic acid complexes known can be viewed in |
|
||||
the NDB. |
|
|
|
|
|
DNA |
SYNTHESIS |
|
|
|
|
Our ability to manufacture specific DNA sequences in almost any desired amounts is well |
|
||||
developed. Nucleic acid chemists have long learned and practiced the powerful approach |
|
||||
of combining chemical and enzymatic syntheses to accomplish their aims. Automated in- |
|
||||
struments exist that perform stepwise chemical synthesis of short DNA strands (oligonu- |
|
||||
cleotides) principally by |
the phosphoramidite method. Synthesis proceeds |
from the 3 |
- |
||
end of the desired sequence using an immobilized nucleotide as the starting material (Fig. |
|
||||
1.6a ). To this are added, successively, the |
desired nucleotides in a blocked, activated |
|
|||
form. After each condenses with the end of the growing chain, it is deblocked to allow the |
|
||||
next step to proceed. It is a routine procedure to synthesize several compounds 20 nu- |
|
||||
cleotides long in a day. Recently instruments have been developed that allow almost a |
|
||||
hundred compounds to be made simultaneously. Typical instruments produce about a |
|
||||
thousand times the amount of material needed for most |
biological experiments. The cost |
|
|||
is about $0.50 to $1.00 per nucleotide in relatively efficient settings. This arises primarily |
|
||||
from the costs of the chemicals needed for the synthesis. Scaling down the process will |
|
||||
reduce the cost accordingly, and efforts to do this are a subject of intense interest. For cer- |
|
||||
tain strategies of large-scale DNA analysis, large |
numbers of different oligonucleotides |
|
|||
are required. The resulting cost will be a significant factor in evaluating the merits of the |
|
||||
overall scheme. The currently used synthetic schemes make it very easy to incorporate |
|
||||
unusual or modified nucleotides at desired places in the sequence, if appropriate deriva- |
|
||||
tives |
are available. They |
also make it very easy to |
add, at the ends |
of the DNA strand, |
|
other functionalities like chemically reactive alkyl amino or thiol groups or useful biolog-
ical ligands like biotin, digoxigenin, or fluorescein. Such derivatives have important uses in many analytical application, as we will demonstrate later.
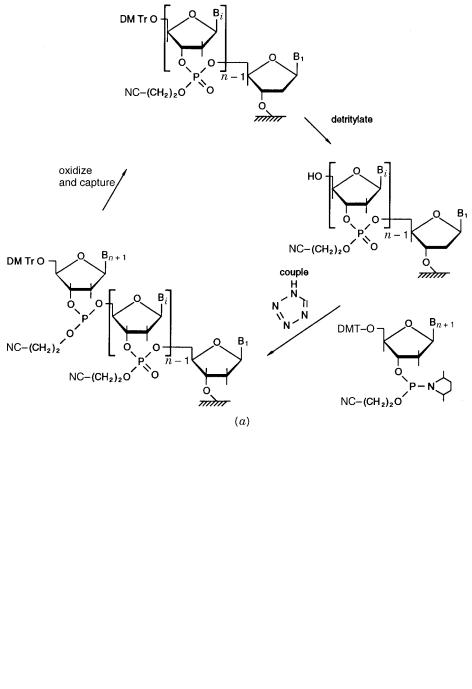
DNA SYNTHESIS |
9 |
Figure 1.6 |
DNA synthesis by combined chemical and enzymatic procedures. |
(a) |
Phos- |
phoramidite chemistry for automated solid state synthesis of DNA chains. |
(b) Assembly of sepa- |
|
|
rately synthesized chains by physical duplex formation and enzymatic joining using DNA ligase. |
|
|
|

10 |
DNA CHEMISTRY AND BIOLOGY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BOX 1.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SIMPLE |
ENZYMATIC MANIPULATION OF |
DNAs |
|
|
|
|
|
|
|
|
|
|
|
|
|
The structure of a DNA strand is an alternating polymer of phosphate and sugar-based |
|
|
|
||||||||||||
units called nucleosides. Thus the ends of the chain can occur at phosphates (p) or at |
|
|
|
||||||||||||
sugar hydroxyls (OH). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Polynucleotide kinase can specifically add a phosphate to the 5 |
|
|
-end of |
a DNA |
|||||||||||
chain. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 HO–ApTpCpG–OH 3 |
|
|
9 |
ATP |
: 5 pApTpCpG–OH 3 |
|
|
|||||||
|
|
|
kinase |
|
|||||||||||
Phosphatases remove phosphates from one or both ends. |
|
|
|
|
|
|
|
|
|
||||||
|
5 pApTpCpGp 3 |
|
|
9 9 |
|
|
|
: 5 HO–ApTpCpG–OH 3 |
|
|
|||||
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
alkaline |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
phosphatase |
|
|
|
|
|
|
|
|
|
|
DNA ligases will join together two DNA strands that lie adjacent along a comple- |
|
|
|
||||||||||||
mentary template. These enzymes require that one of the strands have a 5 |
|
|
-phosphate: |
||||||||||||
|
HO p |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 GpCpCpT GpTpCpCpA |
3 |
|
|
: |
5 GpCpCpTpGpTpCpCpA 3 |
|
|
|||||||
|
3 CpGpGpApCpApGpGpA 5 |
|
|
|
3 CpGpGpApCpApGpGpA 5 |
|
|
||||||||
DNA ligase can also fuse two double-stranded DNAs at their ends provided that 5 |
|
|
- |
||||||||||||
phosphates are present: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 ——— 3 |
|
|
5 p ——— 3 |
|
|
|
|
|
5 ——— p ——— 3 |
|
|
|||
|
3 ——— p5 |
|
3 ——— 5 |
: |
3 ——— p ——— 5 |
|
|
||||||||
This reaction is called blunt-end ligation. It is not particularly sensitive to the DNA |
|
|
|
||||||||||||
sequences of the two reactants. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restriction endonucleases cleave both strands of DNA at or near the site of a spe- |
|
|
|
||||||||||||
cific sequence. They usually cleave at all sites with this particular sequence. The prod- |
|
|
|
||||||||||||
ucts can have blunt-ends, 3 |
-overhangs, or 5 |
|
|
|
-overhangs, as |
shown by the |
examples |
|
|||||||
below: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 —pApCpGpTp—3 |
|
|
|
: 5 —pApC–OH |
|
pGpTp—3 |
|
|
||||||
|
3 —pTpGpCpAp—5 |
|
|
|
3 —pTpGp |
|
|
HO–CpAp—5 |
|
|
|||||
|
5 —pApCpGpTp—3 |
|
|
|
: 5 —pApCpGpT–OH |
|
p—3 |
|
|||||||
|
3 —pTpGpCpAp—5 |
|
|
|
3 —p |
|
|
|
|
|
HO–TpGpCpAp—5 |
|
|
||
|
5 —pApCpGpTp—3 |
|
|
|
: 5 —OH |
|
|
pApCpGpT—3 |
|
|
|||||
|
3 —pTpGpCpAp—5 |
|
|
|
3 —pTpGpCpAp |
HO—5 |
|
|
|||||||
Restriction enzymes always leave 5 |
|
|
|
-phosphates on the cut strands. The resulting |
|
||||||||||
fragments are potential substrates for DNA ligases. Most restriction enzymes cleave at |
|
|
|
||||||||||||
sites with C2 symmetry like the examples shown above. |
|
|
|
|
|
|
|
|
|
||||||
(continued)