

Часть II. Типовые патологические процессы
:судистого свертывания крови независимо от s структурных перестроек, которые могут заекать тромботический процесс. При травматическом повреждении сосуда эомбоз начинается с адгезии тромбоцитов к |стку деэндотелизации. Последняя включает рв этапа: 1) активацию тромбоцитарной мемб-члы; 2) фиксацию активированных тромбоци-вш к галактозиловым группам молекулы кол-ьгена; 3) сокращение тромбоцитов с появлениях псевдоподии.
Активация тромбоцитов является сложным таболическим процессом, связанным с хими-кой модификацией тромбоцитарных мембран аддукцией в них фермента гликозилтрансфе-оы, который взаимодействует со специфически рецептором на молекуле коллагена и обес-чивает тем самым «посадку» тромбоцита на Иввдотелий. Наряду с гликозилтрансферазой активи-тся и другие мембраносвязанные ферменты, астности фосфолипаза А2, обладающая наи-лыпей аффинностью по отношению к фосфа-виэтаноламину. Гидролиз последнего запус-I каскад реакций, включающих высвобож-яе арахидоновой кислоты и последующее об-жание из нее под действием фермента цик-еназы короткоживущих циклических Ююерекисей PGN2 и PGG,,, трансформирую-и под влиянием фермента тромбоксансин-вы в один из самых мощных индукторов аг-ши тромбоцитов и вазоконстрикторов - тром-Ъкгая А,.
Активированный тромбоцит представляет со-своеобразную «пулю», нацеленную на деэн-вввированный участок. Достигнув этого уча-тотчас распластывается на коллагене и екает псевдоподии. Однако для успешной ковки» тромбоцита с коллагеном необходи-сзательное присутствие фактора Виллебран-\зменного фибронектина. В отсутствие белков адгезия не происходит. )ормация тромбоцитов, посаженных на ген, и изменение их формы являются тиле сократительным процессом. В нем при-г участие весь тот набор сократительных ав актомиозинового комплекса, который >ует сокращение гладкомышечных кле-
тевая роль в сократительном акте уделя-i транспорту Са2+ из плотной тубулярной
[ПАТОФИЗИОЛОГИЯ ПЕРИФЕРИЧЕСКОГО
системы, являющейся эквивалентом саркоплаз-матического ретикулума мышц, в цитоплазму. Другие кальциевые пулы, а их в тромбоцитах четыре (плотная тубулярная система, цитоплазма, плотные тельца и митохондрии), не принимают участия в этом процессе. Регуляция транспорта Са2+ осуществляется Са2+-связывающими белками, из которых наиболее значительную роль играет кальмодулин.
Для выполнения транспортной функции Са2+-связывающие белки должны находиться в фос-форилированном состоянии. Фосфорилирование внутриклеточных белков регулируется протеин-киназами циклического 3',5'-аденозинмонофос-фата (цАМФ). Следовательно, транспорт Са2+ и сокращение тромбоцитов тесным образом связаны с внутриклеточным содержанием цАМФ. Снижение в тромбоцитах уровня цАМФ является одним из ранних критериев нарушения функционального состояния тромбоцитов.
Неблагоприятные последствия аккумуляции внутриклеточного Са2+ в сократившихся тромбоцитах связаны по меньшей мере с четырьмя эффектами: 1) разрывом микротрубочек, выполняющих в тромбоците функцию цитоскелета; 2) активацией гуанилатциклазы и усилением синтеза циклического 3', 5'ГМФ, обладающего про-агрегирующим действием; 3) повышением активности фосфолипазы А2 с последующей генерацией тромбоксана А2 и 4) индукцией фосфолипазы С, «нарабатывающей» продукты фосфо-инозитидного обмена (1,2-диацилглицерол, фос-фатидная и лизофосфатидная кислоты), повышающие агрегационную способность тромбоцитов как зависимым, так и независимым от тромбоксана А,, путем.
Адгезия тромбоцитов к субэндотелию является, по существу, I стадией на пути формирования артериального тромбоза. Вслед за ней наступает II стадия агрегации тромбоцитов, состоящая, в свою очередь, из двух последовательных фаз. Первая фаза характеризуется деграну-ляцией и выбросом из тромбоцитов содержимого плотных телец (АДФ, АТФ, АМФ, Фн, адреналина, норадреналина, серотонина, гистамина, ионов Са2+ и др.), вторая - содержимого а-гра-нул (лизосомальные ферменты и др.). При этом мембраны депонирующих гранул сливаются с плазматической мембраной и мембраной каналь-цевой системы тромбоцитов, связанных с поверхностью, в результате чего образуется брешь, че-
195
(ОРГАННОГО) КРОВООБРАЩЕНИЯ
рез которую содержимое органелл выбрасывается наружу.
Появление в кровотоке компонентов тромбо-цитарных гранул приводит к активации соседних интактных тромбоцитов, приклеиванию их друг к другу и к поверхности адгезированных клеток, а в конечном счете - к формированию крупных агрегатов, составляющих основу тром-боцитарного тромба. Одновременно возникает спазм сосуда, вызванный локальным выделением тромбоксана А2 и других вазоактивных веществ.
На практике наибольшее диагностическое значение для выявления повышенной агрегации придается высвобождению в кровоток компонентов плотных телец: АДФ, серотонина, Р-тром-боглобулина и 4-го фактора.
Важную роль играет и уровень 3-го тромбо-цитарного фактора, отражающего изменения
топографии мембранных фосфолипидов, экспозицию на поверхности тех из них, которые в норме находятся в «глубине» плазматической мембраны. К ним, в частности, относится фос-фатидилсерин, мигрирующий на наружную поверхность мембраны тромбоцитов взамен сфин-гомиелина по типу «флип-флоп».
Агрегация тромбоцитов не развивается в отсутствие внеклеточного Са2+, фибриногена и белка, природа которого пока не выяснена. Последний, в частности, отсутствует в плазме крови больных тромбастенией Гланцмана.
Заключительная стадия тромбогенеза связана с активацией контактных факторов плазменного гемостаза, которые адсорбируются на поверхности агрегированных тромбоцитов и запускают «внутренний каскад» свертывания крови, завершающийся выпадением нитей стабилизированного фибрина и консолидацией тромба.
196
гаку также способствует снижение фибрино-гтнческой активации крови, ответственной за .жзис фибриновых сгустков.
Наряду с «внутренним каскадом» в процесс 1ромбообразования включается и «внешний каскад» свертывания крови, связанный с высвобож-евием тканевого тромбопластина.
Активация плазменного гемостаза может воз-жкать и при отсутствии контактных факторов. >этом случае тромбоциты сами запускают «внут-еиний каскад» путем взаимодействия экспони-«■анного на их поверхности V фактора с Характером плазмы, который быстро катализиру-: древращение протромбина в тромбин. Таким образом, тромбоциты выполняют уни-Еюьную роль поверхности, связывающей два хновных звена процесса внутрисосудистого тромбообразования - агрегацию и выпадение сгустка фибрина.
Важно учитывать, что образование полимеров горина в артериальной циркуляции всегда ог-ьннчено и происходит дистальнее тромбоцитар-н «головки» тромба. Это объясняется высокой яростью кровотока, облегчающей разведение ■даление активированных белков свертывания, вторые обеспечивают достаточную доставку гнбиторов свертывания - антитромбина III, фактора гепарина II, протеинов Сив. Помимо тромбоцитов в образовании внутри-■даистых тромбов принимают участие и дру-тки крови, в частности эритроциты и социты. Способность указанных клеток к оукции тромботического процесса связана не -гъко с пассивным захватом их фибриновой яыо, но и активным воздействием на гемоста-й процесс. Последнее особенно наглядно яется при гемолизе эритроцитов, фовождающемся обильным «наводнением» шаатмы АДФ и развитием необратимой агрегату тромбоцитов.
1ередко причиной развития артериального
5оза являются эритроцитоз, приводящий к
жашчению вязкости крови и застою ее в систе-
«шкроциркуляции, сфероцитоз и серповид-
очная анемия, при которой закупорка
ких сосудов может произойти вследствие
гаи эритроцитами эластичности и деформи-
хгги, необходимых для преодоления сопро-
;ения в системе мелких сосудов. Имеются
ьзательства того, что эритроциты в силу круп-
азмеров оттесняют циркулирующие рядом
с ними в потоке крови тромбоциты к периферии и облегчают адгезию последних к субэндотелию.
Роль лейкоцитов в механизмах тромбообра-зования изучена менее подробно, однако известно, что в лейкоцитах активно синтезируются продукты липоксигенного пути метаболизма ара-хидоновой кислоты, и в частности, лейкотрие-ны, которые способны оказывать существенное влияние на активность тромбоцитарной тромбок-сансинтетазы с образованием тромбоксана А2. К тому же в нейтрофилах и других клетках грану-лоцитарного ряда синтезируется тромбоцитакти-вирующий фактор, который тоже может стимулировать повышенную агрегацию тромбоцитов и развитие артериального тромбоза.
Из других внутриклеточных компонентов лейкоцитов, высвобождение которых при острых или хронических воспалительных процессах, а также сепсисе, способно активировать циркулирующие в крови интактные тромбоциты и запускать внутрисосудистую агрегацию, наибольшее значение имеют супероксидные и гидро-ксильные анионрадикалы, лизосомальные гидролазы, ферменты, расщепляющие гепарин, про-теиназы типа нейтрофилина и др.
К тромбогенным компонентам лимфоцитов относятся лимфокины, высвобождающиеся, к примеру, из Т-эффекторов при реакциях замедленного типа.
Принципиальная схема механизма тромбооб-разования в артериях представлена на рис. 46.
8.5.2. Механизмы тромбообразования в венах
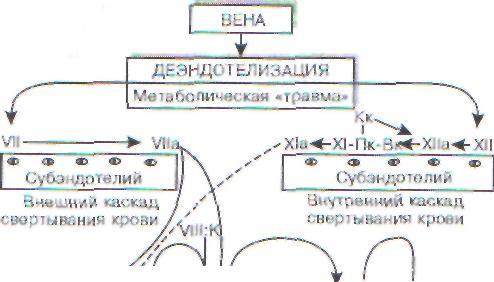
Венозные тромбозы возникают в результате активации плазменного звена гемостаза, что существенно отличает их от артериальных тромбозов, развивающихся на почве сосудисто-тром-боцитарных конфликтов.
Активации плазменного гемостаза в венах благоприятствует гемодинамическая ситуация, создающаяся вблизи венозных клапанов и в местах бифуркаций с замедленным турбулентным потоком крови. Именно в этих «критических» областях возникают ситуации, способствующие адсорбции контактных факторов (XII фактор Хагемана, высокомолекулярный кининоген, пре-калликреин и XI фактор) на отрицательно заряженных структурах обнаженного субэндотелия и запуску внутреннего каскада свертывания
/ ПАТОФИЗИОЛОГИЯ ПЕРИФЕРИЧЕСКОГО (ОРГАННОГО) КРОВООБРАЩЕНИЯ
197
крови. При этом активированный XII фактор расщепляет расположенный рядом прекаллик-реин, связанный с кининогеном, превращая его в калликреин, и активирует фактор XI. Последний, в свою очередь, активирует IX фактор, который взаимодействует с активированным фактором Vlllca. Образующийся комплекс VHIca-IXa расщепляет и активирует ближайшие молекулы фактора X, связанного с фосфолипидами тромбоцитов через остатки у-карбоксиглютама-та. В дальнейшем фактор Ха фиксируется на поверхности тромбоцита и присоединяет молекулы активированного фактора V (Va). Молекулы фактора V либо адсорбируются тромбоцитами из плазмы и затем активируются тромбоци-тарными протеазами, либо высвобождаются в активированной форме из сс-гранул.
На поверхности тромбоцитов комплексы Ха-Va соседствуют с молекулами протромбина (фак-
![]()
Фибрин- Фибри-мономер ноген
Фибрин-полимер
М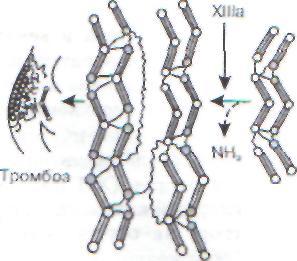 амбраносвязанные
реакцииXIII
Са,
1< "
амбраносвязанные
реакцииXIII
Са,
1< "
Рис. 47. Патогенез венозного тромб 198
тор II). Под влиянием фактора Ха протромбин внутри этого комплекса расщепляется на две части. Первая содержит все остатки у-карбок-сиглютамата и может в течение некоторого времени оставаться в связанном с тромбоцитами состоянии. Вторая часть поступает в кровоток (тромбин, Па). На заключительном этапе тромбин отщепляет 2 пептида от молекулы фибриногена и превращает его сначала в мономерную форму фибрина, а затем - в полимерную. Образуется типичный венозный тромб, стабилизированный нитями полимеризованного фибрина.
Тромбин и полимеры фибрина могут генерироваться также и внешним путем, который инициируется поступающими в кровоток фосфо-липопротеидными мембранами разрушенных клеток и тканей. Последние связывают через Са2+-мостики у-карбоксиглютаматиые остатки витамин К-зависимого профермента фактора VII и превращают его в активный фактор Vila. Одновременно на поверхности мембран эндотелио-цитов активируются факторы X, V и II со всеми вытекающими последствиями.
Процесс формирования тромбиновых масс в венозной системе лимитируется системой ингибиторов свертывания крови. К ним помимо плаз-мина можно отнести антитромбин III - белок, связывающий факторы ХНа, Х1а, Ха, 1Ха и Па, кофактор II гепарина (специфический белковый ингибитор тромбина), протеины С и S.
В норме молекулы антитромбина III присутствуют в циркулирующей крови и связываются с поверхностью эндотелиальных клеток через гепаран-сульфатированный мукополисахарид. Последний участвует также в инактивации тромбина кофактором II гепарина.
Протеины С и S являются витамин К-зависи-мыми белками, циркулирующими в крови и связывающимися через остатки у-карбоксиглютама-та и Са2+ с мембранами эндотелиоцитов и тромбоцитов (протеин S). Физиологическая роль этих белков заключается в том, что они участвуют в инактивации Villa и Va. Механизм инактивации включает связывание тромбина специфическим рецептором тромбомодулином, локализованным на мембране эндотелиоцитов, и активацию комплексом тромбин-тромбомодулин циркулирующего в крови протеина С. Далее активная молекула протеина С соединяется в крови с молекулой протеина S и образуется комплекс, вызывающий протеолиз и элиминацию активирован-