

Biomedical EPR Part-B Methodology Instrumentation and Dynamics - Sandra R. Eaton
.pdf100 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
nitroxyl spin-labels, the  or
or 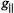 component of the
component of the 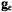 tensor is critical for selection of molecular orientation. In the case of
tensor is critical for selection of molecular orientation. In the case of 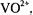 setting
setting 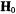 to the –7/2 parallel component of the EPR spectrum (cf., Fig. 1) selects those molecules for which the V = O bond or
to the –7/2 parallel component of the EPR spectrum (cf., Fig. 1) selects those molecules for which the V = O bond or 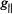 component of the
component of the 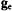 tensor is parallel to the laboratory magnetic field, i.e., the equatorial x,y-plane is perpendicular to
tensor is parallel to the laboratory magnetic field, i.e., the equatorial x,y-plane is perpendicular to 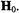 Similarly for spin-labels, setting
Similarly for spin-labels, setting  to the low-field component of the EPR spectrum (cf., Fig. 2) selects those molecules for which the
to the low-field component of the EPR spectrum (cf., Fig. 2) selects those molecules for which the 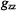 component of the
component of the 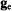 tensor is parallel to
tensor is parallel to 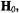 i.e., the molecular x,y-plane is similarly perpendicular to
i.e., the molecular x,y-plane is similarly perpendicular to 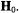 Correspondingly, setting
Correspondingly, setting  to the –3/2 perpendicular component of the EPR spectrum of
to the –3/2 perpendicular component of the EPR spectrum of 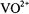 (cf., Fig. 1) selects molecules for which the x,y-molecular plane is parallel to the laboratory field. In the case of nitroxyl spin-labels, the central prominent absorption feature arises from molecules of all orientations, including those for which the molecular plane is perpendicular to
(cf., Fig. 1) selects molecules for which the x,y-molecular plane is parallel to the laboratory field. In the case of nitroxyl spin-labels, the central prominent absorption feature arises from molecules of all orientations, including those for which the molecular plane is perpendicular to 
Figs. 3 and 4 compare the expected pattern of observed ENDOR splittings as a function of the positions of protons and the orientation of 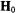 with respect to
with respect to 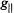 of
of 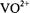 or with respect to
or with respect to 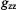 of spin-labels, respectively. When
of spin-labels, respectively. When  is perpendicular to the molecular plane,
is perpendicular to the molecular plane, 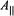 of a proton located along the symmetry axis or
of a proton located along the symmetry axis or 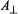 of a proton located near or in the x,y-plane will be observed. On the other hand, if
of a proton located near or in the x,y-plane will be observed. On the other hand, if 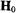 is parallel to the x,y-plane,
is parallel to the x,y-plane, 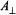 of a proton located along the symmetry axis and both
of a proton located along the symmetry axis and both  and
and  of a proton located near or in the x,y-plane are observed. On this basis, the ENDOR splittings for both types of paramagnetic probes can be classified into three categories:
of a proton located near or in the x,y-plane are observed. On this basis, the ENDOR splittings for both types of paramagnetic probes can be classified into three categories:
Figure 3. Schematic illustration of the relationships of the symmetry axis of the 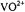 ion to the principal axes of the g tensor and of the hf tensors of nearby protons. Left diagram: Direction of the molecular axes with respect to the V=O bond. Central diagram: Relative positions of protons near the molecular x,y-plane and near or along the z-axis. Each circle represents an orientation of
ion to the principal axes of the g tensor and of the hf tensors of nearby protons. Left diagram: Direction of the molecular axes with respect to the V=O bond. Central diagram: Relative positions of protons near the molecular x,y-plane and near or along the z-axis. Each circle represents an orientation of 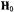 within the g-axis system. Right diagram: Principal hfc components that are detected for equatorially or axially positioned protons according to whether
within the g-axis system. Right diagram: Principal hfc components that are detected for equatorially or axially positioned protons according to whether 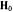 is aligned parallel or perpendicular to the z-axis.
is aligned parallel or perpendicular to the z-axis.

ANGLE-SELECTED ENDOR |
101 |
Figure 4._Magnetic interactions governing angle-selected ENDOR in a nitroxyl spin-label. Upper left-hand portion: Relationships of molecular axes to the principal axes ofthe 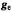 tensor of the nitroxyl group and of the A tensors of nearby protons located in the x,y-plane or on the z axis. Lower left-hand portion: Frozen solution EPR spectrum of a nitroxyl spin-label indicating
tensor of the nitroxyl group and of the A tensors of nearby protons located in the x,y-plane or on the z axis. Lower left-hand portion: Frozen solution EPR spectrum of a nitroxyl spin-label indicating 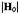 settings (A, B) for microwave saturation. (cf., Fig. 2). Right-hand portion: ENDOR spectra of N-(2,2,5,5-tetramethyl-1-oxypyrrolinyl-3-carbonyl)-L-alaninate enriched with deuterium except at
settings (A, B) for microwave saturation. (cf., Fig. 2). Right-hand portion: ENDOR spectra of N-(2,2,5,5-tetramethyl-1-oxypyrrolinyl-3-carbonyl)-L-alaninate enriched with deuterium except at  recorded for settings A and B of the static magnetic field
recorded for settings A and B of the static magnetic field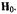 The abscissa measures the observed ENDOR shift (observed frequency of resonance minus the proton Larmor frequency). Reprinted from Mustafi et al. (2001) with permission.
The abscissa measures the observed ENDOR shift (observed frequency of resonance minus the proton Larmor frequency). Reprinted from Mustafi et al. (2001) with permission.
i.splittings that are observed only for the parallel orientation 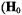 parallel to the
parallel to the  or
or  axis);
axis);
ii.splittings that are observed only for the perpendicular orientation
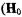 perpendicular to
perpendicular to  and
and
iii. splittings that are observed as common to both parallel and perpendicular orientations.
When g-anisotropy is small compared to the average g value, the maximum hf interaction energy occurs for a field oriented along the
electron-nucleus vector |
and the minimum occurs when the field is |
|||
in the |
For a proton in the x,y-plane, well resolved features corre- |
|||
sponding to |
are observed which persist with an essentially constant |
|||
ENDOR shift at all |
values (Hurst et al., 1985). This observation is an |
|||
important diagnostic feature since |
the observed |
couplings reach a |
||
maximum splitting dependent on |
(Yim and Makinen, 1986; Mustafi et |
|||
al., 1990b). Both |
and |
are observed when the magnetic field orientation |
||
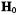 is near or in the molecular x,y-plane. This relationship provides the basis to analyze hf interactions in terms of nuclear coordinates for both types of paramagnetic species. For
is near or in the molecular x,y-plane. This relationship provides the basis to analyze hf interactions in terms of nuclear coordinates for both types of paramagnetic species. For 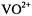 and nitroxyl spin-labels, we have observed hitherto only axially symmetric hf interactions such that each class of
and nitroxyl spin-labels, we have observed hitherto only axially symmetric hf interactions such that each class of
protons gives rise to only |
and |
couplings. Furthermore, the principal |
|
axes of the hf and |
tensors |
are observed to be coincident. These |
|
102 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
relationships may not always obtain. Surprisingly, in the case of 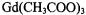 in frozen glassy solutions, the covalent protons of the acetate ligand exhibit axially symmetric hf couplings while the protons of innershell coordinated water molecules within the same complex do not (Yim and Makinen, 1986). Similar observations have been made by others (Gochev and Yordanov, 1993; Zdravkova and Yordanov, 1994).
in frozen glassy solutions, the covalent protons of the acetate ligand exhibit axially symmetric hf couplings while the protons of innershell coordinated water molecules within the same complex do not (Yim and Makinen, 1986). Similar observations have been made by others (Gochev and Yordanov, 1993; Zdravkova and Yordanov, 1994).
3.ENDOR CHARACTERIZATION OF STRUCTURED SOLVENT IN SMALL MOLECULE COMPLEXES AND IN PROTEINS
3.1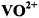 as a Structural Probe of
as a Structural Probe of 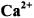 and
and  Sites in Proteins and Nucleotides
Sites in Proteins and Nucleotides
The molecular structure of 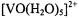 was first elucidated through single crystal ENDOR studies (Atherton and Shackleton, 1980, 1984). However, results of early magnetic resonance studies supported the intuitive expectation that the penta-aquo vanadyl cation
was first elucidated through single crystal ENDOR studies (Atherton and Shackleton, 1980, 1984). However, results of early magnetic resonance studies supported the intuitive expectation that the penta-aquo vanadyl cation 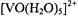 in solution would exhibit square pyramidal geometry with tetragonal symmetry, as had already been shown by X-ray for bis(acetylacetonato)oxovanadium(IV) (Dodge et al., 1961). The results of early nuclear magnetic resonance (NMR) studies using
in solution would exhibit square pyramidal geometry with tetragonal symmetry, as had already been shown by X-ray for bis(acetylacetonato)oxovanadium(IV) (Dodge et al., 1961). The results of early nuclear magnetic resonance (NMR) studies using 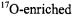 water were consistent with a square pyramidal complex; however, the presence of the fifth axial ligand could not be unambiguously established (Wuthrich and Connick, 1968). Albanese and Chasteen (1978) analyzed the EPR spectrum of
water were consistent with a square pyramidal complex; however, the presence of the fifth axial ligand could not be unambiguously established (Wuthrich and Connick, 1968). Albanese and Chasteen (1978) analyzed the EPR spectrum of  in frozen aqueous medium and were the first to show quantitatively that the dipolar broadening produced by protons of inner sphere coordinated water molecules were consistent with
in frozen aqueous medium and were the first to show quantitatively that the dipolar broadening produced by protons of inner sphere coordinated water molecules were consistent with  as a complex of square pyramidal geometry, using vanadium-oxygen bond distances and valence angles determined crystallographically (Ballhausen et al., 1968). From proton ENDOR spectra of
as a complex of square pyramidal geometry, using vanadium-oxygen bond distances and valence angles determined crystallographically (Ballhausen et al., 1968). From proton ENDOR spectra of 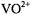 incorporated into host single crystals of
incorporated into host single crystals of 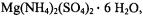 in which the
in which the 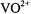 ion replaced
ion replaced 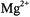 plus one water molecule, Atherton and Shackleton (1980, 1984) determined the principal hfc components for all ten protons of the
plus one water molecule, Atherton and Shackleton (1980, 1984) determined the principal hfc components for all ten protons of the 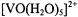 species. They showed that the traceless components of the principal hf tensors are nearly axially symmetric, expected for point-dipole interactions. Because the anisotropy of the various magnetic interactions was explored by rotation about crystal axes with respect to the applied magnetic field
species. They showed that the traceless components of the principal hf tensors are nearly axially symmetric, expected for point-dipole interactions. Because the anisotropy of the various magnetic interactions was explored by rotation about crystal axes with respect to the applied magnetic field  coordinates of each proton in the crystal could be assigned.
coordinates of each proton in the crystal could be assigned.
A more extensive study of the solvation structure of the 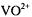 ion was made by ENDOR spectroscopy of frozen solutions of
ion was made by ENDOR spectroscopy of frozen solutions of  in methanol and
in methanol and
ANGLE-SELECTED ENDOR |
|
103 |
water-methanol mixtures on the basis of both and |
ENDOR (Mustafi |
|
and Makinen, 1988). In this study the structure of solvated |
was |
|
assigned through molecular modeling constrained by ENDOR-determined electron-nucleus distances. Although resonance features arising separately from methyl and hydroxyl protons overlap under these conditions, a total of seven pairs of resonance features due to hydroxyl protons with five pairs due to methyl protons were identified by selective deuteration. The principal hfc components for each class of protons were identified from ENDOR spectra with  settings at the –7/2 parallel and –3/2 perpendicular EPR absorption features (cf., Fig. 1), corresponding to magnetic field orientations parallel and perpendicular to the V=O bond, respectively.
settings at the –7/2 parallel and –3/2 perpendicular EPR absorption features (cf., Fig. 1), corresponding to magnetic field orientations parallel and perpendicular to the V=O bond, respectively.
For each class of protons, the principal hfc components for the  complex were axially symmetric. In Table 2 are listed the dipolar and isotropic hfc components, the metal-nucleus distances estimated
complex were axially symmetric. In Table 2 are listed the dipolar and isotropic hfc components, the metal-nucleus distances estimated
according to Eq. (4), and brief comments for each class of nuclei to indicate
the structural relationships of the ligand to |
the |
ion. Because the |
unpaired electron is localized to the metal |
orbital (Ballhausen and Gray, |
|
1962), the isotropic contributions of ligand nuclei in the equatorial plane are significantly higher than for the axially coordinated ligand. Nevertheless, the values of 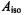 from this study correspond very closely to values determined on the basis of single crystal ENDOR studies (Atherton and
from this study correspond very closely to values determined on the basis of single crystal ENDOR studies (Atherton and
Shackleton, 1980). |
|
Fig. 5 illustrates the solvation structure of |
with ENDOR |
assigned innerand outer-sphere coordinated methanol molecules (Mustafi and Makinen, 1988). In this structure, the outer-sphere coordinated molecules were assigned orientations that could account for plausible hydrogen-bonding interactions with inner-sphere coordinated methanol molecules but which were compatible with the ENDOR-determined vanadium-nucleus distances in axial or equatorial oositions. It was shown by analysis of ENDOR spectra that 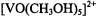 was a unique complex formed only in neat methanol. In water-methanol mixtures, two types of species were identified: one with axially coordinated water trans to the vanadyl oxygen and the other with axially coordinated methanol. Both types of complexes were shown to have only equatorially coordinated water
was a unique complex formed only in neat methanol. In water-methanol mixtures, two types of species were identified: one with axially coordinated water trans to the vanadyl oxygen and the other with axially coordinated methanol. Both types of complexes were shown to have only equatorially coordinated water
molecules. The coordination geometry of |
in neat methanol |
|
and of |
and of |
in water-methanol |
cosolvent mixtures was best accounted for as square-pyramidal with tetragonal symmetry. The structural detail obtained in this ENDOR study approached the precision associated with small molecule X-ray crystallographic studies.

104 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
As shown in Fig. 1, inhomogeneous broadening observed in the spectrum of  provides a particularly pertinent example of the difficulty to assign coordination environment on the basis of EPR alone. Axial and equatorial ligands do not make equivalent contributions to the shf broadening. In the case of
provides a particularly pertinent example of the difficulty to assign coordination environment on the basis of EPR alone. Axial and equatorial ligands do not make equivalent contributions to the shf broadening. In the case of  Albanese and Chasteen (1978) were the first to point out that shf broadening by protons of axial ligands is weak compared to that of equatorial ligands. We have, furthermore, observed that the shf contributions of axial ligands can be masked by equatorial ligands. For instance, the line widths of EPR spectra of nucleotide complexes of
Albanese and Chasteen (1978) were the first to point out that shf broadening by protons of axial ligands is weak compared to that of equatorial ligands. We have, furthermore, observed that the shf contributions of axial ligands can be masked by equatorial ligands. For instance, the line widths of EPR spectra of nucleotide complexes of 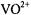 formed with ADP (Mustafi et al., 1992) or 5'- GMP (Jiang and Makinen, 1995) and having
formed with ADP (Mustafi et al., 1992) or 5'- GMP (Jiang and Makinen, 1995) and having  composition are insensitive to exchange of perdeuterated solvent. Nonetheless, for such complexes, in which the nucleotide phosphate groups are equatorially
composition are insensitive to exchange of perdeuterated solvent. Nonetheless, for such complexes, in which the nucleotide phosphate groups are equatorially

ANGLE-SELECTED ENDOR |
105 |
coordinated to 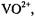 the presence of an axially coordinated solvent molecule could be demonstrated by ENDOR. Similarly, Schweiger and coworkers were able to demonstrate on the basis of
the presence of an axially coordinated solvent molecule could be demonstrated by ENDOR. Similarly, Schweiger and coworkers were able to demonstrate on the basis of  ESEEM that benzaldehyde was axially coordinated through its carbonyl oxygen to
ESEEM that benzaldehyde was axially coordinated through its carbonyl oxygen to 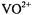 in bis(1R-3- heptafluorobutyrylcamphorate)oxovanadium(IV), a Diels-Alder catalyst (Togni et al., 1993). By applying
in bis(1R-3- heptafluorobutyrylcamphorate)oxovanadium(IV), a Diels-Alder catalyst (Togni et al., 1993). By applying  to the –7/2 parallel EPR absorption feature in pulsed ESEEM studies, they were able to show that the
to the –7/2 parallel EPR absorption feature in pulsed ESEEM studies, they were able to show that the 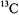 hfc components of an axial aldehyde group were very similar to those of
hfc components of an axial aldehyde group were very similar to those of 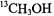 axially coordinated to
axially coordinated to 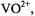 as illustrated in Fig. 5.
as illustrated in Fig. 5.
Figure 5. Stereo diagram of the coordination structure of 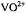 in methanol determined on the basis of ENDOR and molecular modeling. The upper diagram illustrates the complex in stick skeletal form. Broken lines connect the inner-sphere methanol molecules coordinated to the vanadium. The lower diagram illustrates the complex in space-filling form (in the same projection as in the upper diagram), and was drawn to scale for van der Waals radii of 1.53 Å (C), 1.4 Å (O), 1.2 Å (H), and 1.35 Å (V). From Mustafi and Makinen (1988) with permission of the American Chemical Society.
in methanol determined on the basis of ENDOR and molecular modeling. The upper diagram illustrates the complex in stick skeletal form. Broken lines connect the inner-sphere methanol molecules coordinated to the vanadium. The lower diagram illustrates the complex in space-filling form (in the same projection as in the upper diagram), and was drawn to scale for van der Waals radii of 1.53 Å (C), 1.4 Å (O), 1.2 Å (H), and 1.35 Å (V). From Mustafi and Makinen (1988) with permission of the American Chemical Society.
The 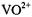 ion occupies a position between
ion occupies a position between 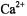 and
and 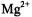 according to ionic charge density defined on the basis of Z/R, where Z is the number of electrons and R the ionic radius (Williams, 1985). This characteristic undoubtedly accounts for its successful application as a paramagnetic probe
according to ionic charge density defined on the basis of Z/R, where Z is the number of electrons and R the ionic radius (Williams, 1985). This characteristic undoubtedly accounts for its successful application as a paramagnetic probe
of |
and |
sites in proteins and nucleotides although |
||
has been used as a substitute also for |
and |
sites in proteins |
||
(Chasteen, 1981, |
1983,1990). We have found |
to |
be a particularly |
|
useful probe of |
sites in proteins, revealing the chemical origins |
|||
of both equatorial and axial ligands (Mustafi and Nakagawa, 1994, 1996; Mustafi et al., 2000). In these studies the resonance features of 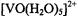 in solution have provided an important basis for analyzing coordination environment in proteins. We have shown that the X-ray structure of
in solution have provided an important basis for analyzing coordination environment in proteins. We have shown that the X-ray structure of 
|
generally considered a model compound of |
|
residues in proteins (Zell et al., 1985), does not account |
for the |
properties of the A and B isoforms of mammalian |
106 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
|
nephrocalcin. This protein containing four distinct |
sites is the |
|
important factor secreted into renal tubules retarding stone formation in the mammalian kidney. Isoforms A and B, exhibiting the tightest 
affinity |
contain 3-4 equivalents of Gla residues |
each while |
isoforms C and D have none and are associated with lower |
|
|
affinity by two orders of magnitude (Nakagawa et al., 1981, 1983, 1985). |
||
Fig. |
6 compares the proton ENDOR spectra of |
of |
nephrocalcin isoforms B and D in natural and perdeuterated aqueous buffer. In each panel, the ENDOR spectrum of  is also compared with that of the nephrocalcin complex. The results show that in isoform B, as in isoform A (Mustafi et al., 2000), only protein residues with nonexchangeable hydrogens are detected as ligands with complete exclusion of solvent water from the inner coordination sphere of the metal ion. The coordination geometry suggested by the
is also compared with that of the nephrocalcin complex. The results show that in isoform B, as in isoform A (Mustafi et al., 2000), only protein residues with nonexchangeable hydrogens are detected as ligands with complete exclusion of solvent water from the inner coordination sphere of the metal ion. The coordination geometry suggested by the  complex places the Gla residues as bidentate ligands in the equatorial plane with an
complex places the Gla residues as bidentate ligands in the equatorial plane with an
axial |
water (Zell et al., 1985). Clearly, the structure of the |
|
complex cannot account for the coordination environment of |
the |
sites in nephrocalcin isoforms A and B. For isoforms C and D, on |
the other hand, two water molecules are detected by ENDOR in the inner coordination sphere of the metal ion (Mustafi et al., 2000), but these isoforms do not contain Gla residues.
Nephrocalcin isoforms A and B undergo a more prominent conformational change upon binding  or
or  than do isoforms C and D, as measured through circular dichroism (Mustafi and Nakagawa, 1994, 1996; Mustafi et al., 2000). Under the assumption that the interior regions of isoforms A and B acquire a lower dielectric constant through the conformational change, becoming less polar, it is unusual that water is
than do isoforms C and D, as measured through circular dichroism (Mustafi and Nakagawa, 1994, 1996; Mustafi et al., 2000). Under the assumption that the interior regions of isoforms A and B acquire a lower dielectric constant through the conformational change, becoming less polar, it is unusual that water is
excluded from the inner coordination sphere of the bound |
ions. It is of |
||
interest that parvalbumin contains two |
sites, of which the high |
||
affinity site |
similarly has no inner-sphere coordinated water |
||
(Kretsinger |
and Nockolds, 1973). |
Since nephrocalcin |
hinders the |
aggregation and growth of microcrystals of calcium oxalate into renal stones, elucidating the molecular basis by which stone formation is inhibited presents a challenging problem.
Since |
binds only to the phosphate groups of nucleotides (Happe and |
|
Morales, |
1966) and is generally required for enzyme-catalyzed phosphoryl |
|
transfer reactions in cells, it is important to determine structures of |
||
|
complexes or to employ spectroscopic probes that closely |
|
simulate |
interactions with phosphate groups. In our laboratory we have |
|
observed through EPR and ENDOR spectroscopy that |
is coordinated |
|
only to the phosphate groups of nucleotides, similar to |
that observed for |
|
|
complexes (Mustafi et al., 1992; Jiang and Makinen, 1995). |
|
ANGLE-SELECTED ENDOR |
107 |
Since it was possible to demonstrate in these studies that  binding was inhibited by
binding was inhibited by  with no evidence of non-specific binding of the
with no evidence of non-specific binding of the 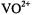 ion to the ribose hydroxyl groups or to the base hetero-atoms, it is likely that
ion to the ribose hydroxyl groups or to the base hetero-atoms, it is likely that 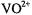 may serve as the most suitable paramagnetic probe to simulate
may serve as the most suitable paramagnetic probe to simulate 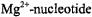 interactions in spectroscopic studies.
interactions in spectroscopic studies.
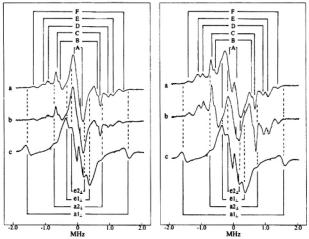
Figure 6. Proton ENDOR spectra of 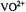 complexes in frozen aqueous solutions. In the lefthand panel, spectra correspond to the following: a,
complexes in frozen aqueous solutions. In the lefthand panel, spectra correspond to the following: a, 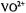 : nephrocalcin isoform B (4:1 molar ratio) in protiated aqueous buffer; b,
: nephrocalcin isoform B (4:1 molar ratio) in protiated aqueous buffer; b,  : isoform B (4:1 molar ratio) in deuterated aqueous buffer; and c,
: isoform B (4:1 molar ratio) in deuterated aqueous buffer; and c,  complex. In the right-hand panel, the spectra correspond to the same conditions but for nephrocalcin isoform D. In both sets of ENDOR spectra, the magnetic field was set to –3/2 perpendicular EPR absorption feature, and proton ENDOR absorptions from inner-sphere coordinated water molecules in axial and equatorial positions, labeled
complex. In the right-hand panel, the spectra correspond to the same conditions but for nephrocalcin isoform D. In both sets of ENDOR spectra, the magnetic field was set to –3/2 perpendicular EPR absorption feature, and proton ENDOR absorptions from inner-sphere coordinated water molecules in axial and equatorial positions, labeled  and
and 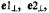 respectively, are identified by stick diagrams. For the complex of isoform B, no resonance features for axial or equatorial coordinated water molecules are detected, as indicated by the broken vertical lines. For the
respectively, are identified by stick diagrams. For the complex of isoform B, no resonance features for axial or equatorial coordinated water molecules are detected, as indicated by the broken vertical lines. For the 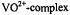 of isoform D, proton ENDOR features for inner shell equatorial water molecules are seen, as indicated by the solid vertical lines between spectra a and b. In each panel ENDOR line pairs deriving from amino acid residues of the protein, labeled A-F, are indicated by stick diagrams with solid vertical lines for spectra a. The ENDOR line pairs are equally spaced about the free proton Larmor frequency of 13.9 MHz. The abscissa indicates the ENDOR shift. Reprinted from Mustafi et al. (2000) with permission of the CMB Association.
of isoform D, proton ENDOR features for inner shell equatorial water molecules are seen, as indicated by the solid vertical lines between spectra a and b. In each panel ENDOR line pairs deriving from amino acid residues of the protein, labeled A-F, are indicated by stick diagrams with solid vertical lines for spectra a. The ENDOR line pairs are equally spaced about the free proton Larmor frequency of 13.9 MHz. The abscissa indicates the ENDOR shift. Reprinted from Mustafi et al. (2000) with permission of the CMB Association.
Of the five types of nucleic acid bases in DNA and RNA, guanine is unique because its nucleosides and nucleotides are capable of forming selfstructured assemblies in solution through hydrogen-bonding to yield G – G base pairs and G-quartets. The latter are square planar arrays of the guanine bases with Hoogsteen hydrogen bonding interactions with each other. These assemblies are important because the 3’-overhang regions of DNA strands in the cell are rich in guanine and serve as the point of attachment of the DNA strand to protein subunits comprising the mitotic spindle apparatus in cell

108 DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN
division (Blackburn, 2000). It is thought that G-quartet formation in these regions of DNA strands may be important in binding to the protein.
It has long been known that G-quartet assemblies can be formed from 5’- GMP in solution as a square-planar array of hydrogen-bonded guanine bases (Gellert et al., 1962). Equilibria controlling the formation of G-quartets and of stacked quartets to form octets and higher order assemblies are sensitive to pH and the presence of sodium and potassium ions (Pinavaia et al., 1978). Although helical fibers of stacked quartets of 5'-GMP were characterized crystallographically (Zimmerman, 1976) and by infrared spectroscopy (Audet et al., 1991), the pucker of the ribose ring and conformation of the base moiety in these helical arrays were not established.
Jiang and Makinen (1995) demonstrated on the basis of NMR studies that the 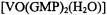 complex in the presence of excess 5’-GMP enters into self-structured quartet and octet assemblies through hydrogen-bonding like the free nucleotide. They were able to show that the ENDOR shifts of the protons assigned to the ribose moiety and to the 8-H position in the nucleic acid base were identical for the monomeric form of
complex in the presence of excess 5’-GMP enters into self-structured quartet and octet assemblies through hydrogen-bonding like the free nucleotide. They were able to show that the ENDOR shifts of the protons assigned to the ribose moiety and to the 8-H position in the nucleic acid base were identical for the monomeric form of 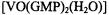 as well as for the metal-nucleotide complex incorporated into quartet and octet assemblies. This observation indicated that the conformation of the metal-nucleotide complex was unchanged upon its incorporation into G-quartet assemblies.
as well as for the metal-nucleotide complex incorporated into quartet and octet assemblies. This observation indicated that the conformation of the metal-nucleotide complex was unchanged upon its incorporation into G-quartet assemblies.
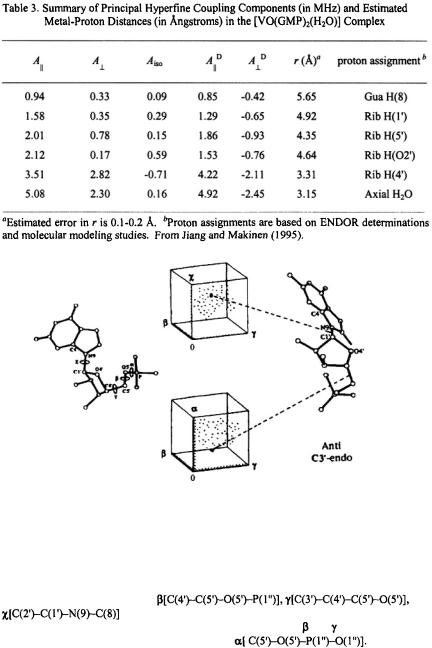
Table 3 summarizes the ENDOR transition frequencies and their assignments for the 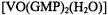 complex in solution. Fig. 7 illustrates the results of torsion angle search calculations. Only a small family of conformations accommodate the ENDOR-determined vanadiumproton distances within van der Waals hard-sphere constraints. Different conformations of the ribose ring were tested. The ENDOR determined electron-proton distances restricted the ribose conformation to a C3'- endo pucker. Modeling studies using the ENDOR distances as constraints were able to rule out the C2’- endo conformation, which is the other prevalent conformation found for monomeric ribonucleotides (Saenger, 1984), On the other hand, for G-quartets formed with
complex in solution. Fig. 7 illustrates the results of torsion angle search calculations. Only a small family of conformations accommodate the ENDOR-determined vanadiumproton distances within van der Waals hard-sphere constraints. Different conformations of the ribose ring were tested. The ENDOR determined electron-proton distances restricted the ribose conformation to a C3'- endo pucker. Modeling studies using the ENDOR distances as constraints were able to rule out the C2’- endo conformation, which is the other prevalent conformation found for monomeric ribonucleotides (Saenger, 1984), On the other hand, for G-quartets formed with 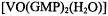 in solution, the guanine base was restricted to the anti conformation. The X-ray structure of double-stranded
in solution, the guanine base was restricted to the anti conformation. The X-ray structure of double-stranded 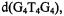 which forms hydrogen-bonded arrays of G- quartets in crystals, shows the guanine base to occupy both syn and anti conformations within each planar quartet array (Kang et al., 1992). However, the quality of the electron density map did not allow an unambiguous assignment of ribose pucker. In deoxyribonucleotides the absence of the 2’-hydroxyl group allows more conformational flexibility with respect to ring pucker than in ribonucleotides. The structure of the
which forms hydrogen-bonded arrays of G- quartets in crystals, shows the guanine base to occupy both syn and anti conformations within each planar quartet array (Kang et al., 1992). However, the quality of the electron density map did not allow an unambiguous assignment of ribose pucker. In deoxyribonucleotides the absence of the 2’-hydroxyl group allows more conformational flexibility with respect to ring pucker than in ribonucleotides. The structure of the
complex incorporated into a G-quartet, as derived
ANGLE-SELECTED ENDOR |
109 |
through EPR, ENDOR, and NMR investigations (Jiang and Makinen, 1995) is illustrated in Fig. 8.
Figure 7. Angle maps showing conformational space accessible to the 5’-GMP moiety in  under van der Waals hard-sphere constraints only (low density dots) and upon application of the distance constraints in Table 3 (high density dots connected by broken lines to the conformer accommodating vanadium-proton distance constraints within their line width based uncertainties). Because of the closed ring structures of the nucleic acid base and of the ribose, only four dihedral angles define the conformation of 5’-GMP (standard designations shown in the left-hand structure). The right-hand structure depicts the ENDOR assigned conformation as C3’- endo. In the upper angle map, the axes correspond to 0–360°
under van der Waals hard-sphere constraints only (low density dots) and upon application of the distance constraints in Table 3 (high density dots connected by broken lines to the conformer accommodating vanadium-proton distance constraints within their line width based uncertainties). Because of the closed ring structures of the nucleic acid base and of the ribose, only four dihedral angles define the conformation of 5’-GMP (standard designations shown in the left-hand structure). The right-hand structure depicts the ENDOR assigned conformation as C3’- endo. In the upper angle map, the axes correspond to 0–360°
of rotation for the dihedral angles |
and |
over which the search calculations were carried out in 1° |
|
increments. The lower angle map shares axes for dihedral angles |
and while the third axis |
represents 0–360° of rotation for the dihedral angle |
|