

Biomedical EPR Part-B Methodology Instrumentation and Dynamics - Sandra R. Eaton
.pdfEPR INTERFACED TO RAPID MIXING |
69 |
Figure 11. The EPR spectra of spin labeled yeast iso-1-cytochrome c in 0.1 M pH 5 sodium acetate buffer. (A) Folded protein at 15 °C. (B) First integral of A. (C) Denatured protein in 2 M GdnHCl at 15 °C. (D) C102-SL at 75 °C. (E) Comparison of native and GdnHCldenatured C102-SL at the same system gain. Figure reprinted with permission from Qu et al. (1997); Copyright 1997 American Chemical Society.
Figure 12. The kinetic unfolding transient EPR signal following 1:1 mixing of 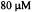 C102SL in 0.1 M phosphate buffer with 4 M GdnHCl denaturant pH 6.5, 2 °C. The central H(0) EPR feature was monitored. The data presented is the result of 2 summed transients. Inset shows fit of kinetic transient to single exponential with unfolding rate constant
C102SL in 0.1 M phosphate buffer with 4 M GdnHCl denaturant pH 6.5, 2 °C. The central H(0) EPR feature was monitored. The data presented is the result of 2 summed transients. Inset shows fit of kinetic transient to single exponential with unfolding rate constant 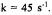 This experiment required less than
This experiment required less than 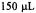 of
of 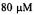 spin labeled protein. Figure reprinted with permission from Qu et al. (1997); Copyright 1997 American Chemical Society.
spin labeled protein. Figure reprinted with permission from Qu et al. (1997); Copyright 1997 American Chemical Society.
70 |
CHARLES P. SCHOLES |
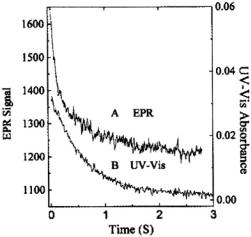
The more complex refolding kinetics of our labeled cytochrome were studied by stopped-flow EPR (Figure 13A at pH 6.5). The spin probe signal at pH 6.5 (and 5.0) showed a fast kinetic process (~100 ms times or faster at 2 °C), compatible with the time range over which hydrogen/deuterium exchange sensed early helix formation of the C & N helices (Roder et al., 1988; Elöve et al., 1994; Sosnick et al., 1994). At pH 6.5 an additional slower kinetic phase (~0.5 s) was reported by the spin probe attached at C102. Heme-ligation-sensitive UV-Vis absorption spectroscopy exclusively indicated this slower (0.5-1 s) folding (Figure 13B), a folding attributed to recovery from the kinetic trap of pH-dependent heme-histidine mis-ligation (Sosnick et al., 1994; Colón et al., 1997).
Figure 13. This figures provides a comparison of the EPR and UV-Vis stopped-flow kinetics of protein refolding where initially 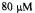 unfolded C102-SL in pH 6.5, 1.8 M GdnHCl was mixed 1:1 with pH 6.5, 0.1 M phosphate buffer at 2 °C. (A) The EPR trace exhibiting biphasic behavior with rates of 12 and
unfolded C102-SL in pH 6.5, 1.8 M GdnHCl was mixed 1:1 with pH 6.5, 0.1 M phosphate buffer at 2 °C. (A) The EPR trace exhibiting biphasic behavior with rates of 12 and  (B) The UV-Vis kinetic trace obtained at 407 nm exhibiting a slower biphasic behavior with rates of 1.8 and
(B) The UV-Vis kinetic trace obtained at 407 nm exhibiting a slower biphasic behavior with rates of 1.8 and 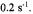 Figure reprinted with permission from Qu et al. (1997); Copyright 1997 American Chemical Society.
Figure reprinted with permission from Qu et al. (1997); Copyright 1997 American Chemical Society.
The work of Qu et al. (1997) showed evidence of a faster probe immobilization and an incipient “burst” of folding that occurred in a time less than the ~7 ms dead time of the 1997-vintage grid-mixer stopped-flow apparatus. To resolve such a faster refolding process, a micro ball-mixer with submicroliter dead volume was subsequently integrated with the dielectric resonator (Grigoryants et al., 2000). Thermal melting characterization made us aware that spin labeling the C102 position had led to a 20 °C lower melting temperature than that of unlabeled wild type protein (Qu et al., 1997). The location of the C102 sulfur prior to labeling is
EPR INTERFACED TO RAPID MIXING |
71 |
a region of hydrophobic packing (Louie and Brayer, 1990), and so labeling C102 from within that region had destabilized the protein, probably by distorting the hydrophobic packing.
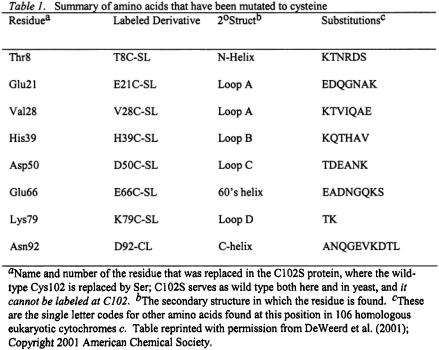
We had noted that the utility of spin labeling had been markedly improved through cysteine-specific spin labeling via methanethiosulfonate (Hubbell and Altenbach, 1994; Hubbell et al., 1998; Hubbell et al., 2000), and our next goal was to attach MTSSL in new cysteine-mutated sites. The new labeling sites (See Figure 9 and Table 1) were selected as external, hydrophilic, mutation-tolerant sites. These sites, when mutated to cysteine and spin labeled, were less perturbing to folding than the naturally occurring C102. Furthermore, labeling at numerous non-perturbing locations presented the possibility of following folding at numerous locations, not just C102. In summary, the initial kinetic work on C102-SL (Qu et al., 1997) put the folding study of spin labeled protein into the framework of other recent folding measurements. It pointed toward a systematic folding study with cysteine-directed mutants, and it pointed toward technical improvements in mixer-resonator design.
72 |
CHARLES P. SCHOLES |
3.3.1Variable Velocity Liquid Flow EPR Applied to Submillisecond Protein Folding
A schematic of Grigoryants’ elegant capillary mixer intimately connected to a mini EPR probe is provided in Figure 14. Liquid to be mixed flowed from two inlet tubes, through a narrow annular slit, past a platinum ballmixer, and into the outlet tube which immediately entered the DR. With this device we followed the submillisecond refolding kinetics of iso-1-cyt c labeled at its at naturally occurring C102.
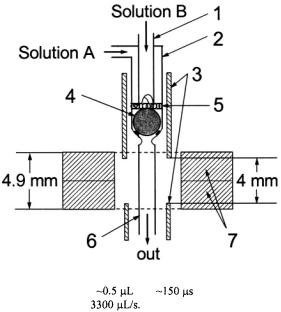
Figure 14. Schematic of the prototype mini ball mixer integrated to a 9.5 GHz dielectric
resonator. The system has a dead volume |
and |
delivery time to center of the |
observation zone with a flow velocity of |
Solutions A and B are two solutions to |
|
be mixed. Parts are as follows: (1) 0.3 mm i.d. inlet capillary; (2) 0.6 mm i.d. inlet capillary;
(3) 0.9 mm i.d. silver shield tubes; (4) 0.57 mm Pt sphere/ball mixer; (5) 0.1 mm frit made from 0.076 mm Pt wire; (6) 0.4 mm i.d. outlet capillary; (7) two ceramic toroids of the dielectric resonator. The height of the dielectric resonator structure is 4.9 mm, and the height of the EPR-active zone is 4 mm. Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
By changing the programmed flow velocity, we changed the delivering time of mixed liquids from the mixing point at the ball mixer to the zone of observation (i.e., the dielectric resonator). It was necessary first to calibrate the time resolution of the instrument; the calibration process also led to an empirical estimate of the dead volume and gave insight into the mixing efficiency. The timing calibration of the instrument was performed by using the rapid destruction of TEMPO (2,2,6,6-tetramethyl-4-piperidinol) by
EPR INTERFACED TO RAPID MIXING |
73 |
sodium dithionite, a reaction which was initially determined by stopped-flow EPR to exhibit fast linear decay. The fast linear decay time meant that during flow there was a meaningful and detectable change in the EPR signal as a function of the flow velocity. For the TEMPO-dithionite reaction the decay of signal intensity depended linearly on the delivery (dead) time between mixing and the center of the EPR-active zone and inversely upon the flow velocity. In Figure 15 we present a plot of the EPR signal amplitude obtained during flow versus the inverse of the flow velocity. This plot was also made versus the delivery time which was the age of the sample computed from the linearly decaying amplitude of the EPR signal (and not from a pre-conceived concept of dead volume). At all but the slowest velocity the signal amplitude scaled linearly with the inverse of flow velocity. The implication of this linear scaling was that the actual time for turbulent mixing in the narrow annular region near the ball (Regenfuss et al., 1985) was much less than the dead time for the sample to flow from mixer to the EPR-active volume, at all but the slowest velocity. This meant that the time-zero for the start of folding is set at the mixer. Thus the subsequent time for flow from the mixer to the center of the EPR-active zone is the time for folding kinetics and not for a complex compendium of solution mixing and folding kinetics. The dead volume obtained from the decay rate of the calibrating chemical reaction and the known fluid flow rate was about 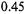
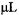 (Grigoryants et al., 2000), a volume which was virtually identical to the liquid volume from the middle of the ball to the middle of the DR. At the slowest velocity
(Grigoryants et al., 2000), a volume which was virtually identical to the liquid volume from the middle of the ball to the middle of the DR. At the slowest velocity 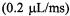 and longest delivery times (>2 ms) there was evidence for inefficient mixing since the signal during flow deviated in a positive direction from linear decay. That deviation implied that at low fluid velocity the time for turbulent mixing was not much less than the time for the sample to flow from the mixing element to the EPR-active volume. Fortunately, kinetics in the >2 ms time regime can be independently obtained by the stopped-flow method with the velocity before stopping
and longest delivery times (>2 ms) there was evidence for inefficient mixing since the signal during flow deviated in a positive direction from linear decay. That deviation implied that at low fluid velocity the time for turbulent mixing was not much less than the time for the sample to flow from the mixing element to the EPR-active volume. Fortunately, kinetics in the >2 ms time regime can be independently obtained by the stopped-flow method with the velocity before stopping 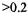
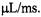 The limitation on the fastest flow was the high pressure needed to achieve that flow combined with the stress tolerance of our thick-walled (presently 4.6 mm i.d., 30 mm o.d.) glass driver syringes; we refrained from flowing at a faster rate than 3 mL/s.
The limitation on the fastest flow was the high pressure needed to achieve that flow combined with the stress tolerance of our thick-walled (presently 4.6 mm i.d., 30 mm o.d.) glass driver syringes; we refrained from flowing at a faster rate than 3 mL/s.
We have so far used solutions whose viscosity is about that of water (0.01 poise). Conceivably more viscous solvents than the solutions used here could lead to a more inefficient, less turbulent mixing and to the need for higher flow velocity to induce efficient, more turbulent mixing. A Reynolds number approach to turbulence (see Ch. 41 in Feynman et al. (1964)) indicates that turbulence increases with flow velocity and decreases with viscosity. With higher flow velocity would come the higher RAM pressures which could endanger the glass driver syringes.
74 |
CHARLES P. SCHOLES |
Figure 15. Presents the velocity flow/time calibration curve for our micro ball mixer. We show the dependence of the intensity of the continuously flowing EPR signal upon the reciprocal of the flow velocity for the mixing of 2 mM TEMPO with 30 mM Na-dithionite contained in 0.1 M  buffer, and we show the time scale that we calibrated from the linear decay of the dithionite-TEMPO reaction for delivery of solutions from mixing to the center of the EPR-active zone. Evidence for inefficient mixing was observed at the slowest flow rate (i.e., flow velocity
buffer, and we show the time scale that we calibrated from the linear decay of the dithionite-TEMPO reaction for delivery of solutions from mixing to the center of the EPR-active zone. Evidence for inefficient mixing was observed at the slowest flow rate (i.e., flow velocity 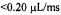 or reciprocal velocity
or reciprocal velocity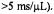 Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
This “Variation of Flow Velocity” method here allowed us initially to reach a kinetic resolution time of 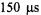 (and most recently,
(and most recently,  two orders faster than the former stopped-fiow method (Qu et al., 1997). Direct evidence was provided (Figure 16) for a burst of folding or protein compacting at C102 that occurred within
two orders faster than the former stopped-fiow method (Qu et al., 1997). Direct evidence was provided (Figure 16) for a burst of folding or protein compacting at C102 that occurred within 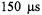 of mixing at 20 °C and within
of mixing at 20 °C and within 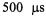 of mixing at 7 °C. There clearly was a temperature dependence to this burst, implying an activation energy to the early submillisecond folding process at C102-SL. The reactant usage for the overall set of experiments which involved separate measurements at different flow velocities was 2 mL of
of mixing at 7 °C. There clearly was a temperature dependence to this burst, implying an activation energy to the early submillisecond folding process at C102-SL. The reactant usage for the overall set of experiments which involved separate measurements at different flow velocities was 2 mL of  spin labeled C102SL. The ball-mixer device also served nicely as a highly efficient stopped-flow probe with total reactant usage of about
spin labeled C102SL. The ball-mixer device also served nicely as a highly efficient stopped-flow probe with total reactant usage of about 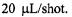 Replacing the grid mixer enabled us in stopped-flow mode to obtain folding times at room temperature below 10 ms as shown in Figure 17. The ball mixer device is the first ever created to give submillisecond time resolution with welldefined dead times in EPR flow mode measurements, and the technology opens a new area to biophysics for the kinetic study of radicals. Development to decrease the dead volume further, to computerize the
Replacing the grid mixer enabled us in stopped-flow mode to obtain folding times at room temperature below 10 ms as shown in Figure 17. The ball mixer device is the first ever created to give submillisecond time resolution with welldefined dead times in EPR flow mode measurements, and the technology opens a new area to biophysics for the kinetic study of radicals. Development to decrease the dead volume further, to computerize the
EPR INTERFACED TO RAPID MIXING |
75 |
system so that the velocity may be continuously swept, and to obtain overall rapidly scanned spectra during flow is underway.
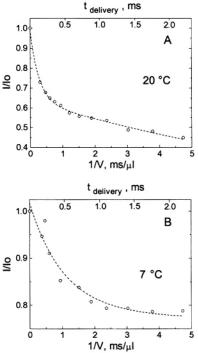
Figure 16. (A) shows the decreasing amplitude of the EPR signal at 20 °C represented by the fractional signal intensity 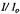 (shown with open circles) of the central EPR feature. 0.4 mM spin labeled C102-SL in 0.8 M GdnHCl was mixed 1:1 with dilute 0.1 M Na acetate buffer to obtain a 0.4 M GdnHCl concentration in which the protein was ~70 % refolded.
(shown with open circles) of the central EPR feature. 0.4 mM spin labeled C102-SL in 0.8 M GdnHCl was mixed 1:1 with dilute 0.1 M Na acetate buffer to obtain a 0.4 M GdnHCl concentration in which the protein was ~70 % refolded. 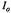 was the initial unfolded derivative EPR signal amplitude, and I was the time-dependent signal amplitude as folding progressed. The decrease of the central EPR feature from folding protein is shown as a function of the inverse of flow velocity (lower horizontal axis) and of the calibrated delivering time (upper horizontal axis) between the mixer and the center of the observation EPR zone. The fit (dashed lines) at 20 °C indicates the presence of two components of the folding processes, one with a 0.12 ± 0.02 ms exponential decay and the other with a 6.2 ± 0.8 ms exponential decay. The former had an amplitude which was ~37 % of the signal change, and the latter had an amplitude which was ~63 % of the signal change. (B) Shows the decreasing fractional amplitude taken at 7 °C, where there was only one component in the 0.1 to 2 ms range whose decay time was 0.5 ± 0.05 ms; this phase accounted for ~25 % of the overall signal change. Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
was the initial unfolded derivative EPR signal amplitude, and I was the time-dependent signal amplitude as folding progressed. The decrease of the central EPR feature from folding protein is shown as a function of the inverse of flow velocity (lower horizontal axis) and of the calibrated delivering time (upper horizontal axis) between the mixer and the center of the observation EPR zone. The fit (dashed lines) at 20 °C indicates the presence of two components of the folding processes, one with a 0.12 ± 0.02 ms exponential decay and the other with a 6.2 ± 0.8 ms exponential decay. The former had an amplitude which was ~37 % of the signal change, and the latter had an amplitude which was ~63 % of the signal change. (B) Shows the decreasing fractional amplitude taken at 7 °C, where there was only one component in the 0.1 to 2 ms range whose decay time was 0.5 ± 0.05 ms; this phase accounted for ~25 % of the overall signal change. Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
76 |
CHARLES P. SCHOLES |
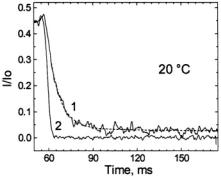
Figure 17. This figure shows the stopped-flow kinetics (curve 1) measured at 20 °C represented by the fractional signal intensity 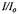 under the same solution mixing conditions for C102-SL as in Figure 16A. In this case there was a rapid transient showing a time decay of 7 ± 2 ms, consistent with the slow 6 ms process measured by flow EPR in Figure 16A. Because there is a finite fast braking time for the flow, we also provide the profile of a very fast reaction (curve 2) of TEMPO with dithionite (1 mM TEMPO + 1 M dithionite) which represents the apparatus slowing function. The experimental apparatus slowing function is shown because we rely on RAM braking and on fluid friction in our narrow tubes rather than a stopping syringe to stop flow. Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
under the same solution mixing conditions for C102-SL as in Figure 16A. In this case there was a rapid transient showing a time decay of 7 ± 2 ms, consistent with the slow 6 ms process measured by flow EPR in Figure 16A. Because there is a finite fast braking time for the flow, we also provide the profile of a very fast reaction (curve 2) of TEMPO with dithionite (1 mM TEMPO + 1 M dithionite) which represents the apparatus slowing function. The experimental apparatus slowing function is shown because we rely on RAM braking and on fluid friction in our narrow tubes rather than a stopping syringe to stop flow. Figure reprinted from Grigoryants et al. (2000) with permission from the Biophysical Society.
3.3.2EPR-detected Folding Kinetics of Externally Located Cysteine-Directed Spin-Labeled Mutants of Iso-1-Cytochrome c
We next systematically probed protein folding in yeast iso-1-cyt c at cysteine-directed spin-labeled locations. The locations studied were not previously directly probed by other techniques, and we observed them on a time scale stretching from 50 microseconds to seconds. The following mutation-tolerant, externally located cysteine labeling sites shown in Fig. 9 above and Table 1 below were chosen (in helices: T8C, E66C, N92C; in loops: E21C, V28C, H39C, D50C, K79C). These externally located spin labels generally retained high local mobility upon the protein to which they were attached. In contrast to labeling at naturally occurring C102, these did not destabilize the fold or perturb the thermal melting temperature (DeWeerd et al., 2001). These mutant yeast cytochromes were created in our lab by our application of recombinant DNA technology. Using a high yield heterologous E. coli plasmid system (Pollock et al., 1998; Morar et al., 1999), we obtained ~100 mg quantities of these mutant cytochromes for kinetic work.
EPR INTERFACED TO RAPID MIXING |
77 |
Dilution of denaturant (i.e., GdnHCl) induced folding, and the folding caused a kinetic change in the spin label EPR signal as folding altered the motion of the spin label. Under folding conditions, including the presence of imidazole to eliminate kinetic trapping due to heme-misligation, a phase of folding on the 20-30 ms time scale at room temperature was everywhere found, as shown in Figure 18. The time constants and amplitudes of folding phases for all spin labeled derivatives were given in Table 2 of DeWeerd et al. (2001). The N-C helical regions, which include T8C-SL and N92C-SL, have been identified by hydrogen exchange measurements as the location of early helix formation occurring on this time scale (Roder et al., 1988; Elöve et al., 1994; Englander et al., 1998). However, all regions of the protein, not just the N & C locale, experienced the folding phase in the 20-30 ms time window as reported by spin labels.
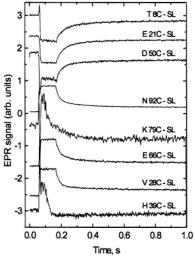
Figure 18. This figure presents refolding behavior as followed by ball mixer stopped-flow EPR over a 1 s range showing fast folding in the 20-30 ms range for the following samples: T8C-SL, E21C-SL, D50C-SL, N92C-SL, K79C-SL, E66C-SL, V28C-SL, and H39C-SL. Initially unfolded protein of approximate 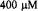 concentration in pH 5.0, 1.7 M GdnHCl, 200 mM imidazole was mixed in a 1:1 fashion with 0.05 M acetate buffer, 200 mM imidazole at pH 5.0 to initiate folding. K79C-SL and H39C-SL required 100 shots at
concentration in pH 5.0, 1.7 M GdnHCl, 200 mM imidazole was mixed in a 1:1 fashion with 0.05 M acetate buffer, 200 mM imidazole at pH 5.0 to initiate folding. K79C-SL and H39C-SL required 100 shots at 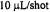 so that the time for flow during a shot was half as long and the flow stopped sooner for these two samples. The other samples required approximately 50 shots at
so that the time for flow during a shot was half as long and the flow stopped sooner for these two samples. The other samples required approximately 50 shots at 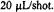 The traces are shown normalized to approximately the same amplitude to make a convenient presentation. The overall amplitudes of the intrinsic recovery signal will vary from one site to the next because the signal difference between folded and unfolded protein will vary from one labeling site to the next. The traces which show the most noise are actually the ones from sites where the difference between folded and unfolded EPR signals was the smallest. Figure reprinted with permission from DeWeerd et al. (2001); Copyright 2001 American Chemical Society.
The traces are shown normalized to approximately the same amplitude to make a convenient presentation. The overall amplitudes of the intrinsic recovery signal will vary from one site to the next because the signal difference between folded and unfolded protein will vary from one labeling site to the next. The traces which show the most noise are actually the ones from sites where the difference between folded and unfolded EPR signals was the smallest. Figure reprinted with permission from DeWeerd et al. (2001); Copyright 2001 American Chemical Society.

78 CHARLES P. SCHOLES
The global nature of the 20-30 ms phase, which our site-directed spin labels reported, is a simple yet important finding for the folding of cytochrome c. A common criticism of spin labels is that they perturb the phenomena that they are supposed to measure; the general evidence of our study is that our spin labeling strategy reported common themes and not highly perturbed, disconnected kinetic events.
Although it was expected and generally found that the derivative signal would become more intense for the unfolded protein because the spin label is more mobile in unfolded protein, there were several instances (T8C-SL, E21C-SL, D50C-SL) where the signal became larger for the folded protein than for the unfolded protein in the time range beyond a millisecond (Figure 18). The paramagnetic ferric heme is itself attached at amino acids 14, 17, and 18. Investigation is underway to determine if for T8C-SL, E21C-SL, D50C-SL it is paramagnetic relaxation in the unfolded state or greater impediment to probe motion in the unfolded state which causes their signals in the time range beyond a millisecond to increase as the protein folds.
The ultrafast submillisecond component reported by flow EPR from D50C-SL showed probe immobilization on the 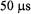 time scale, as indicated in Figure 19, even at 5 °C. This immobilization was an order of magnitude
time scale, as indicated in Figure 19, even at 5 °C. This immobilization was an order of magnitude
faster immobilization than C102-SL showed |
at 7 |
°C) at a |
comparable temperature (Grigoryants et al., 2000). |
It is possible |
that D50 |
lies in or near an initiation site where a loop rapidly forms on the 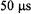 time scale (Hagen et al., 1996); the region where D50 is located is not one where early helical structure occurs (Roder et al., 1988), nor is it in a hydrophobic region. At V28C-SL, H39C-SL, E66C-SL, and K79C-SL as well as D50CSL stopped-flow EPR indicated a substantial percentage of burst phase. Therefore a comprehensive investigation by rapid-mix, submillisecond flow EPR is underway on V28C-SL, H39C-SL, E66C-SL, K79C-SL, and D50CSL to characterize the time scale(s), spatial extent, and activation energy of
time scale (Hagen et al., 1996); the region where D50 is located is not one where early helical structure occurs (Roder et al., 1988), nor is it in a hydrophobic region. At V28C-SL, H39C-SL, E66C-SL, and K79C-SL as well as D50CSL stopped-flow EPR indicated a substantial percentage of burst phase. Therefore a comprehensive investigation by rapid-mix, submillisecond flow EPR is underway on V28C-SL, H39C-SL, E66C-SL, K79C-SL, and D50CSL to characterize the time scale(s), spatial extent, and activation energy of
the submillisecond folding/pre-folding kinetics in the |
ms time |
window. |
|