

Вопрос№5 Энтропия вещества из системы - мера беспорядка расположения в них частиц.
Термодинамическая энтропия S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы.
Понятие
энтропии было впервые введено
в 1865 году Рудольфом
Клаузиусом.
Он определил изменение
энтропии термодинамической
системы при обратимом
процессе как
отношение общего количества
тепла ![]() к
величине абсолютной
температуры
к
величине абсолютной
температуры ![]() :
:
![]() .
.
Например, при температуре 0 °C, вода может находиться в жидком состоянии и при незначительном внешнем воздействии начинает быстро превращаться в лед, выделяя при этом некоторое количество теплоты. При этом температура вещества так и остается 0 °C. Изменяется состояние вещества, сопровождающееся выделением тепла, вследствие изменения структуры.
Рудольф
Клаузиус дал величине ![]() имя
«энтропия», происходящее от греческого
слова τρoπή,
«изменение» (изменение, превращение,
преобразование). Данное равенство
относится к изменению энтропии, не
определяя полностью саму энтропию.
имя
«энтропия», происходящее от греческого
слова τρoπή,
«изменение» (изменение, превращение,
преобразование). Данное равенство
относится к изменению энтропии, не
определяя полностью саму энтропию.
Эта формула применима только для изотермического процесса (происходящего при постоянной температуре). Её обобщение на случай произвольногоквазистатического процесса выглядит так:
![]() ,
,
где ![]() —
приращение (дифференциал) энтропии
некоторой системы, а
—
приращение (дифференциал) энтропии
некоторой системы, а ![]() —
бесконечно малое количество теплоты,
полученное этой системой.
—
бесконечно малое количество теплоты,
полученное этой системой.
Необходимо обратить внимание на то, что рассматриваемое термодинамическое определение применимо только к квазистатическим процессам (состоящим из непрерывно следующих друг за другом состояний равновесия).
Поскольку
энтропия является функцией
состояния,
в левой части равенства стоит её полный
дифференциал.
Напротив, количество теплоты
является функцией
процесса,
в котором эта теплота была передана,
поэтому ![]() считать
полным дифференциалом нельзя.
считать
полным дифференциалом нельзя.
Энтропия, таким образом, согласно вышеописанному, определена вплоть до произвольной аддитивной постоянной. Третье начало термодинамики позволяет определить её точнее: предел величины энтропии равновесной системы при стремлении температуры к абсолютному нулю полагают равным нулю.
Существует мнение, что мы можем смотреть на S и как на меру беспорядка в системе. В определённом смысле это может быть оправдано, потому что мы думаем об «упорядоченных» системах как о системах, имеющих очень малую возможность конфигурирования, а о «беспорядочных» системах как об имеющих очень много возможных состояний. Собственно, это просто переформулированное определение энтропии как числа микросостояний на данное макросостояние.
Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный «беспорядок», так как при этом будут максимальные возможности конфигурирования.
Границы применимости понимания энтропии как меры беспорядка
Подобное определение беспорядка термодинамической системы как количества возможностей конфигурирования системы фактически дословно соответствует определению энтропии как числа микросостояний на данное макросостояние. Проблемы начинаются в двух случаях:
-
когда начинают смешивать различные понимания беспорядка, и энтропия становится мерой беспорядка вообще;
-
когда понятие энтропии применяется для систем, не являющихся термодинамическими.
В обоих этих случаях применение понятия термодинамической энтропии совершенно неправомерно[1].
Рассмотрим оба пункта подробнее.
Рассмотрим пример термодинамической системы — распределение молекул в поле тяготения. В этом случае наиболее вероятным распределением молекул будет распределение согласнобарометрической формуле Больцмана. Другой пример — учёт электромагнитных сил взаимодействия между ионами. В этом случае наиболее вероятным состоянием, соответствующим минимуму свободной энергии, будет упорядоченное кристаллическое состояние, а совсем не «хаос», хотя в состоянии «хаоса» значение конфигурационной энтропии системы и ниже. (Термин «хаос» здесь понимается в смысле беспорядка — в наивном смысле. К хаосу в математическом смысле как сильно неустойчивой нелинейной системе это не имеет отношения, конечно.)
Рассмотрим случай с кристаллической решёткой более подробно. Кристаллическая решётка может быть и в равновесном, и в неравновесном состоянии, как и любая термодинамическая система. Скажем, возьмём следующую модель — совокупность взаимодействующих осцилляторов. Рассмотрим некоторое неравновесное состояние: все осцилляторы имеют одинаковое отклонение от положения равновесия. С течением времени эта система перейдёт в состояние ТД равновесия, в котором отклонения (в каждый момент времени) будут подчинены некоторому распределению типа Максвелла (только это распределение будет для отклонений, и оно будет зависеть от типа взаимодействия осцилляторов). В таком случае максимум энтропии будет действительно реализовывать максимум возможностей конфигурирования, то есть — беспорядок согласно вышеуказанному определению. Но данный «беспорядок» вовсе не соответствует «беспорядку» в каком-либо другом понимании, например, информационному. Такая же ситуация возникает и в примере с кристаллизацией переохлаждённой жидкости, в которой образование структур из «хаотичной» жидкости идёт параллельно с увеличением энтропии.
То есть при образовании кристалла из переохлажденной жидкости энтропия увеличивается с одновременным ростом температуры. Если кристаллизация сопровождается отводом тепла из системы, то энтропия при этом уменьшится.
Это неверное понимание энтропии появилось во время развития теории информации, в связи с парадоксом термодинамики, связанным с мысленным экспериментом т. н. «демона Максвелла». Суть парадокса заключалась в том, что рассматривалось два сосуда с разными температурами, соединённых узкой трубкой с затворками, которыми управлял т. н. «демон». «Демон» мог измерять скорость отдельных летящих молекул, и таким образом избирательно пропускать более быстрые в сосуд с высокой температурой, а более медленные — в сосуд с низкой. Из этого мысленного эксперимента вытекало кажущееся противоречие со вторым началом термодинамики.
Парадокс может быть разрешён при помощи теории информации. Для измерения скорости молекулы «демон» должен был бы получить информацию о её скорости. Но всякое получение информации — материальный процесс, сопровождающийся возрастанием энтропии. Количественный анализ[2] показал, что приращение энтропии при измерении превосходит по абсолютной величине уменьшение энтропии, вызванное перераспределением молекул «демоном».
Вопрос№6 Второй закон термодинамики
Второй Закон Термодинамики, как и Первый (Закон сохранения энергии) установлен эмпирическим путем. Впервые его сформулировал Клаузиус: "теплота сама собой переходит лишь от тела с большей температурой к телу с меньшей температурой и не может самопроизвольно переходить в обратном направлении".
Другая формулировка: все самопроизвольные процессы в природе идут с увеличением энтропии. (Энтропия - мера хаотичности, неупорядоченности системы).
Вопрос №7 Свободная энергия (энергия Гиббса, Гельмгольца) как критерий направления химической реакции и предела её протекания.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.
Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпиисистемы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста её энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж)
Свобо́дная эне́ргия Гельмго́льца (или просто свобо́дная эне́ргия) — термодинамический потенциал, убыль которого в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
Свободная энергия Гельмгольца для системы с постоянным числом частиц определяется так:
-
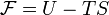 ,
где
,
где 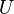 — внутренняя
энергия,
— внутренняя
энергия, 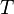 —
абсолютная температура,
—
абсолютная температура,  — энтропия.
— энтропия.
Отсюда дифференциал свободной энергии равен:
-
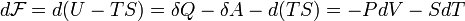 .
.
Видно,
что это выражение является полным
дифференциалом относительно независимых
переменных ![]() и
и ![]() .
Поэтому часто свободную энергию
Гельмгольца для равновесного состояния
выражают как функцию
.
Поэтому часто свободную энергию
Гельмгольца для равновесного состояния
выражают как функцию ![]() .
.
Для системы с переменным числом частиц дифференциал свободной энергии Гельмгольца записывается так:
-
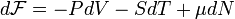 ,
,
где ![]() — химический
потенциал,
а
— химический
потенциал,
а ![]() —
число частиц в системе. При этом свободная
энергия Гельмгольца для равновесного
состояния записывается как функция
—
число частиц в системе. При этом свободная
энергия Гельмгольца для равновесного
состояния записывается как функция ![]() .
.
В соответствии с рекомендациями ИЮПАК энергию Гельмгольца в химической термодинамике можно также обозначать как A[1].
Вопрос№8 Скорость химической реакции и методы её регулирования. Зависимость скорости реакции от природы и концентрации реагентов. Закон действия масс (ЭДМ) для гомогенных и гетерогенных реакций.
Количественной характеристикой того, насколько быстро протекает данная
реакция, является скорость химической реакции, т. е. скорость
расходования реагентов или скорость появления продуктов. При этом
безразлично, о каком из участвующих в реакции веществе идет речь,
поскольку все они связаны между собой через уравнение реакции. По
изменению количества одного из веществ можно судить о
соответствующих изменениях количеств всех остальных.
Скорость реакции определяется изменением молярной концентрации
одного из реагирующих веществ:
V = +-((С2 – С1)/(t2 – t1)) = +-(дельта С /дельта t)
где С1 и С2 - молярные концентрации веществ в моменты времени t1 и t2
соответственно (знак (+) – если скорость определяется по продукту
реакции, знак (–) – по исходному веществу).
Скорость реакции в данном случае обычно выражается в моль/(л•с).
Реакции происходят при столкновении молекул реагирующих веществ. Ее
скорость определяется количеством столкновений и вероятностью того,
что они приведут к превращению. Число столкновений определяется
концентрациями реагирующих веществ, а вероятность реакции - энергией
сталкивающихся молекул.
Скорость гетерогенной реакции выражается в моль/(м2•с).
Чтобы управлять химическими реакциями, важно не только уметь
определять их скорости, но и выяснить, какие условия оказывают на них
влияние. Раздел химии, изучающий скорость химических реакций и
влияние на нее различных факторов, называется химической кинетикой
1) Влияние
концентрации реагентов
Зависимость
скорости химической реакции от
концентрации реагентов выражается законом
действующих масс:
![]() где
k – константа скорости реакции, СА и
СВ –
текущие концентрации реагентов, nA и
nB -
порядок реакции по реагенту А и В
соответственно.
где
k – константа скорости реакции, СА и
СВ –
текущие концентрации реагентов, nA и
nB -
порядок реакции по реагенту А и В
соответственно.
 Анализ
уравнения закона действующих масс
приводит к выводу, что:
1)
скорость химической реакции увеличивается
при увеличении концентрации реагентов;
2)
скорость реакции в большей степени
зависит от концентрации того реагента,
порядок по которому выше;
3)
скорость химической реакции уменьшается
во времени, так как уменьшаются текущие
концентрации реагирующих веществ.
Из
уравнения закона действующих масс
вытекает возможность использования
двух технологических приемов, позволяющих
увеличить скорость химической реакции:
1)
работа с более концентрированным сырьем;
при необходимости сырье предварительно
обогащают, то есть тем или иным способом
увеличивают в нем содержание основного
компонента;
2)
использование избытка одного из
реагентов; что позволяет сохранять в
ходе реакции высокие значения произведения
, а, следовательно, и высокую скорость
процесса.
Анализ
уравнения закона действующих масс
приводит к выводу, что:
1)
скорость химической реакции увеличивается
при увеличении концентрации реагентов;
2)
скорость реакции в большей степени
зависит от концентрации того реагента,
порядок по которому выше;
3)
скорость химической реакции уменьшается
во времени, так как уменьшаются текущие
концентрации реагирующих веществ.
Из
уравнения закона действующих масс
вытекает возможность использования
двух технологических приемов, позволяющих
увеличить скорость химической реакции:
1)
работа с более концентрированным сырьем;
при необходимости сырье предварительно
обогащают, то есть тем или иным способом
увеличивают в нем содержание основного
компонента;
2)
использование избытка одного из
реагентов; что позволяет сохранять в
ходе реакции высокие значения произведения
, а, следовательно, и высокую скорость
процесса.
В избытке берется тот реагент: - порядок по которому выше; - стоимость которого ниже; - отделение которого от продуктов проще.
Избыток
не должен сильно разбавлять реакционную
смесь, нарушать теплофизические свойства
реакционной смеси и тепловое регулирование
реактора. Кроме того, слишком большой
избыток реагента приводит к лишним
экономическим затратам. Величину избытка
определяют экспериментально; обычно
берут избыток реагента 1,7 – 1,8 по сравнению
со стехиометрией.
 Вполне
очевидно, что, чем выше скорость химической
реакции, тем меньше время достижения
одной и той же величины конверсии.
Поэтому при больших начальных, а,
следовательно, и текущих концентрациях
реагента время проведения химической
реакции уменьшается.
Анализ
кривой α = f (τ) также показывает, что
скорость изменения конверсии уменьшается
во времени, и при значениях α, близких
к 100%, изменение конверсии очень
незначительно. Поэтому всегда существует
некоторое оптимальное значение конверсии
(обычно 80-90%), при достижении которого
вести процесс дальше экономически
нецелесообразно. Время достижения этой
величины конверсии называют оптимальным
временем реакции.
2) Влияние
давления
Скорость
реакции зависит от числа столкновений
молекул реагирующих веществ, а число
столкновений зависит в свою очередь от
числа молекул в единице объема, то есть
от концентрации. С увеличением давления
объем вещества уменьшается, и молекулы
теснее располагаются в единице объема,
то есть увеличение давление равнозначно
увеличению концентрации. Особенно
значителен этот эффект у газов. Жидкости
и тем более твердые вещества малосжимаемы
и эффект давления проявляется только
в области сверхдавлений (1000 атм и
выше).
В
случае газофазных реакций концентрации
реагентов в законе действующих масс
можно заменить их парциальными
давлениями.
Вполне
очевидно, что, чем выше скорость химической
реакции, тем меньше время достижения
одной и той же величины конверсии.
Поэтому при больших начальных, а,
следовательно, и текущих концентрациях
реагента время проведения химической
реакции уменьшается.
Анализ
кривой α = f (τ) также показывает, что
скорость изменения конверсии уменьшается
во времени, и при значениях α, близких
к 100%, изменение конверсии очень
незначительно. Поэтому всегда существует
некоторое оптимальное значение конверсии
(обычно 80-90%), при достижении которого
вести процесс дальше экономически
нецелесообразно. Время достижения этой
величины конверсии называют оптимальным
временем реакции.
2) Влияние
давления
Скорость
реакции зависит от числа столкновений
молекул реагирующих веществ, а число
столкновений зависит в свою очередь от
числа молекул в единице объема, то есть
от концентрации. С увеличением давления
объем вещества уменьшается, и молекулы
теснее располагаются в единице объема,
то есть увеличение давление равнозначно
увеличению концентрации. Особенно
значителен этот эффект у газов. Жидкости
и тем более твердые вещества малосжимаемы
и эффект давления проявляется только
в области сверхдавлений (1000 атм и
выше).
В
случае газофазных реакций концентрации
реагентов в законе действующих масс
можно заменить их парциальными
давлениями.![]() Известно,
что парциальные давления компонентов
смеси пропорциональны их мольным
долям.
Известно,
что парциальные давления компонентов
смеси пропорциональны их мольным
долям.
![]() где
р – общее давление, NA и
NB –
мольные доли компонентов А и В. Тогда
где
р – общее давление, NA и
NB –
мольные доли компонентов А и В. Тогда
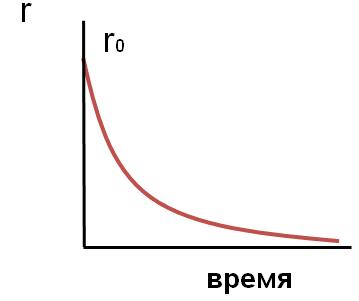
![]() То
есть скорость газофазной реакции
пропорциональна давлению в степени,
равной суммарному порядку реакции.
Изменение давления наиболее сильно
влияет на реакции высокого порядка. При
низком значении порядка реакции повышение
давления вызывает незначительное
изменение скорости процесса.
То
есть скорость газофазной реакции
пропорциональна давлению в степени,
равной суммарному порядку реакции.
Изменение давления наиболее сильно
влияет на реакции высокого порядка. При
низком значении порядка реакции повышение
давления вызывает незначительное
изменение скорости процесса.
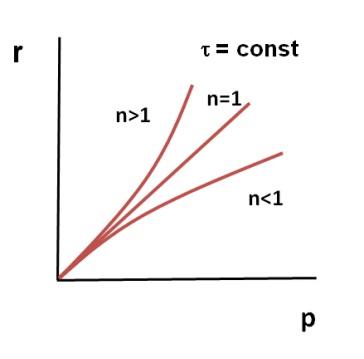
 Зависимость
конверсии от давления показывает, что
существует некоторое оптимальное
значение давления (ропт.),
выше которого рост конверсии при
повышении давления очень незначителен,
и поэтому использование более высоких
давлений становится нецелесообразным.
На
практике использование давления в
качестве фактора управления химической
реакцией ограничено большими затратами
на его реализацию и возможностями
техники высокого давления.
Зависимость
конверсии от давления показывает, что
существует некоторое оптимальное
значение давления (ропт.),
выше которого рост конверсии при
повышении давления очень незначителен,
и поэтому использование более высоких
давлений становится нецелесообразным.
На
практике использование давления в
качестве фактора управления химической
реакцией ограничено большими затратами
на его реализацию и возможностями
техники высокого давления.
Вопрос№9 Зависимость скорости химической реакции от температуры. Правило Вант-Гоффа.
3) Влияние
температуры
Скорость
реакции зависит не только от числа
столкновений реагирующих молекул, но
и от их энергии. Появление активных
молекул, обладающих повышенной реакционной
способностью, является результатом
различных физических воздействий на
вещество. При активации реагентов тем
или иным способом передачи энергии
увеличивается константа скорости
реакции. Способы активации могут быть
разные: фотохимическая, плазмохимическая,
ультразвуковая и др. Наиболее часто
используется термоактивация, то
есть увеличение скорости реакции под
воздействием температуры.
Скорость
химической реакции при увеличении
температуры на 10 градусов возрастает
в 2 - 4 раза. Это эмпирическое правило
называется правилом
Вант-Гоффа.
Оно справедливо в области средних
температур (до 4000С).
Более
точно влияние температуры можно
описать уравнением
Аррениуса:
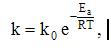 где
Еа –
энергия активации, Т – температура, R –
газовая постоянная, k0 –
предэкспоненциальный множитель,
зависящий только от природы реагирующих
частиц.
где
Еа –
энергия активации, Т – температура, R –
газовая постоянная, k0 –
предэкспоненциальный множитель,
зависящий только от природы реагирующих
частиц.
 Подставим
это уравнение в кинетическое уравнение
и увидим, что скорость химической реакции
при увеличении температуры возрастает
по экспоненте.
Подставим
это уравнение в кинетическое уравнение
и увидим, что скорость химической реакции
при увеличении температуры возрастает
по экспоненте.  Степень
влияния температуры зависит от величины
энергии активации. Реакции с большей
энергией активации более чувствительны
к изменению температуры, реакции с малой
энергией активации нечувствительны.
Каждая
реакция значительно более чувствительна
к изменению температуры при низких
температурах, чем при высоких.
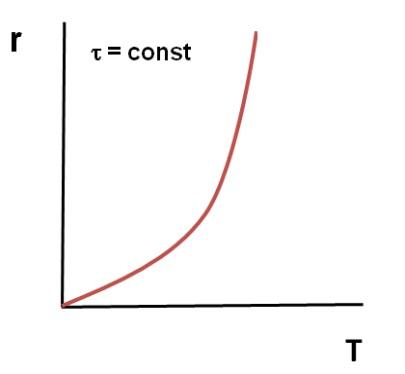
Кривая
зависимости конверсии от температуры
носит S-образный характер.
Степень
влияния температуры зависит от величины
энергии активации. Реакции с большей
энергией активации более чувствительны
к изменению температуры, реакции с малой
энергией активации нечувствительны.
Каждая
реакция значительно более чувствительна
к изменению температуры при низких
температурах, чем при высоких.
Кривая
зависимости конверсии от температуры
носит S-образный характер.
 При
низких температурах мала скорость
процесса, поэтому невелик подъем кривой
α = f (T). По мере повышения температуры
скорость реакции увеличивается по
экспоненте, что приводит к резкому
подъему кривой. Но с увеличением степени
превращения реагентов снижается их
концентрация, а, следовательно, и скорость
процесса, поэтому функциональная
зависимость α = f (T) асимптотически
приближается к единице.
Анализ
кривой на графике приводит к выводу,
что в данном случае тоже существует
некоторое оптимальное значение
задаваемого параметра – температуры,
выше которого вести процесс экономически
нецелесообразно из-за незначительного
увеличения конверсии при высоких
энергетических затратах.
Термический
способ активации химической реакции
очень часто используется в технологии
(до 95% от общего числа промышленных
синтезов). Недостатком этого способа
является высокие энергетические затраты
и низкая селективность (при повышении
температуры ускоряются не только целевые
реакции, но и побочные).
В
технологической практике обычно ставится
задача повышения скорости химической
реакции. Однако необходимо понимать,
что вышеописанные закономерности
управления скоростью химической реакции
можно использовать также и для уменьшения
скорости реакции, например, для подавления
побочных реакций. Иногда
малоконцентрированное сырье или
разбавление реакционной массы
растворителем или инертным газом
используют для уменьшения скорости
целевой реакции. Обычно это связано со
сложностью организации быстрого
теплоотвода в случае сильноэкзотермических
реакций.
При
низких температурах мала скорость
процесса, поэтому невелик подъем кривой
α = f (T). По мере повышения температуры
скорость реакции увеличивается по
экспоненте, что приводит к резкому
подъему кривой. Но с увеличением степени
превращения реагентов снижается их
концентрация, а, следовательно, и скорость
процесса, поэтому функциональная
зависимость α = f (T) асимптотически
приближается к единице.
Анализ
кривой на графике приводит к выводу,
что в данном случае тоже существует
некоторое оптимальное значение
задаваемого параметра – температуры,
выше которого вести процесс экономически
нецелесообразно из-за незначительного
увеличения конверсии при высоких
энергетических затратах.
Термический
способ активации химической реакции
очень часто используется в технологии
(до 95% от общего числа промышленных
синтезов). Недостатком этого способа
является высокие энергетические затраты
и низкая селективность (при повышении
температуры ускоряются не только целевые
реакции, но и побочные).
В
технологической практике обычно ставится
задача повышения скорости химической
реакции. Однако необходимо понимать,
что вышеописанные закономерности
управления скоростью химической реакции
можно использовать также и для уменьшения
скорости реакции, например, для подавления
побочных реакций. Иногда
малоконцентрированное сырье или
разбавление реакционной массы
растворителем или инертным газом
используют для уменьшения скорости
целевой реакции. Обычно это связано со
сложностью организации быстрого
теплоотвода в случае сильноэкзотермических
реакций.
Вопрос№10 Уравнение Аррениуса. Энергия активации. Влияние катализатора на скорость реакции. Механизм действия катализатора.
Уравне́ние
Арре́ниуса устанавливает
зависимость константы
скорости ![]() химической
реакции от температуры
химической
реакции от температуры ![]() .
.
Согласно
простой модели столкновений, химическая
реакция между двумя исходными веществами
может происходить только в результате
столкновения молекул этих
веществ. Но не каждое столкновение ведёт
к химической реакции. Необходимо
преодолеть определённый энергетический
барьер,
чтобы молекулы начали друг с другом
реагировать. То есть молекулы должны
обладать некой минимальной энергией
(энергия
активации ![]() ),
чтобы этот барьер преодолеть.
Из распределения
Больцмана для
кинетической энергии молекул известно,
что число молекул, обладающих энергией
),
чтобы этот барьер преодолеть.
Из распределения
Больцмана для
кинетической энергии молекул известно,
что число молекул, обладающих энергией ![]() ,
пропорционально
,
пропорционально ![]() .
В результате скорость химической реакции
представляется уравнением, которое
было получено шведским химиком Сванте
Аррениусомиз термодинамических соображений:
.
В результате скорость химической реакции
представляется уравнением, которое
было получено шведским химиком Сванте
Аррениусомиз термодинамических соображений:
![]()
Здесь ![]() характеризует
частоту столкновений реагирующих
молекул,
характеризует
частоту столкновений реагирующих
молекул, ![]() — универсальная
газовая постоянная.
— универсальная
газовая постоянная.
В
рамках теории активных соударений ![]() зависит
от температуры, но эта зависимость
достаточно медленная:
зависит
от температуры, но эта зависимость
достаточно медленная:
![]()
Оценки
этого параметра показывают, что изменение
температуры в диапазоне от 200 °C до
300 °C приводит к изменению частоты
столкновений ![]() на
10 %.
на
10 %.
В
рамках теории активированного комплекса
получаются другие зависимости ![]() от
температуры, но во всех случаях более
слабые, чем экспонента.
от
температуры, но во всех случаях более
слабые, чем экспонента.
Уравнение Аррениуса стало одним из основных уравнений химической кинетики, а энергия активации — важной количественной характеристикой реакционной способности веществ.
Энергия активации в элементарных реакциях, минимальная энергия реагентов (атомов, молекул и других частиц), достаточная для того, чтобы они вступили в хим. реакцию, т. е. для преодоления барьера на поверхности потенциальной энергии, отделяющего реагенты от продуктов реакции.
Потенциальный
барьер - максимум потенциальной энергии,
через который должна пройти система в
ходе элементарного акта химического
превращения. Высота потенциального
барьера для любого пути, проходящего
через переходное состояние, равна
потенциальной энергии в переходном
состоянии. Если в сложной реакции,
состоящей из последовательных и
параллельных элементарных реакций,
имеется лимитирующая элементарная
реакция (реакция с максимальным
характерным временем), то ее энергия
активации является и энергией активации
сложной реакции. В макроскопической
химической кинетике энергия активации
- энергетический параметр Еа, входящий
в <i.Аррениуса
уравнение. ![]() где к
- константа
скорости. А -
предэкспоненциальный множитель
(постоянная или слабо зависящая от
температуры величина); k
- константа
Больцмана; Т
- абсолютная
температура. График зависимости lnk от
1/kT (аррениусов
график) - прямая линия. Наблюдаемая
энергия активации вычисляется из
тангенса угла наклона этой прямой. В
общем случае сложных реакций параметр Еа в
уравнении Аррениуса является функцией
энергии активации отдельных стадий, и
определяемая энергия активации называется
эффективной (эмпирической, кажущейся).
где к
- константа
скорости. А -
предэкспоненциальный множитель
(постоянная или слабо зависящая от
температуры величина); k
- константа
Больцмана; Т
- абсолютная
температура. График зависимости lnk от
1/kT (аррениусов
график) - прямая линия. Наблюдаемая
энергия активации вычисляется из
тангенса угла наклона этой прямой. В
общем случае сложных реакций параметр Еа в
уравнении Аррениуса является функцией
энергии активации отдельных стадий, и
определяемая энергия активации называется
эффективной (эмпирической, кажущейся).
Любой процесс,
сопровождающийся каким-либо изменением
энергии, является экзотермическим в
одном направлении и эндотермическим в
другом. Энергия активации экзотермического
и эндотермического направлений реакции,
обозначаемые соотв. ![]() и
и ![]() ,
связаны соотношением:
,
связаны соотношением: ![]()
где Q - .теплота реакции при Т= 0. Качественная одномерная геометрическая иллюстрация связи энергии активации с высотой потенциального барьера и теплотой реакции представлена на рис., где q -координата реакции ; Е1 и Е2 -уровни энергии соответственно основного состояния реагентов и продуктов реакции.
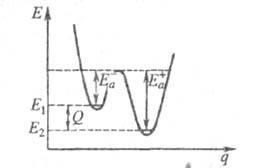
Энергетич. схема элементарной реакции.
Для
реакций рекомбинации свободных
радикалов (в том числе и атомов), а также
для широкого класса экзотермических
ионно-молекулярных реакций энергия
активации равна нулю или очень мала по
сравнению с типичными значениями энергий
хим. связей Есв.
Для реакций, сопровождающихся одновременно
разрывом одних и образованием других
химических связей, ![]() составляет
обычно от сотых до десятых долей Есв,
если среди реагентов есть свободные
радикалы, и сравнима с Есв , если
реагенты - химически насыщенные
молекулы.
составляет
обычно от сотых до десятых долей Есв,
если среди реагентов есть свободные
радикалы, и сравнима с Есв , если
реагенты - химически насыщенные
молекулы. ![]() может
быть аномально большой (например, больше
энергии возбуждения атома Е*)
в реакциях тушения электронного
возбуждения при столкновениях атомов:
А + А
может
быть аномально большой (например, больше
энергии возбуждения атома Е*)
в реакциях тушения электронного
возбуждения при столкновениях атомов:
А + А ![]() А
+ А, если точка пересечения термов
реагентов и продуктов реакции расположена
высоко по сравнению с Е* или
термы не пересекаются.
А
+ А, если точка пересечения термов
реагентов и продуктов реакции расположена
высоко по сравнению с Е* или
термы не пересекаются.
Влияние катализатора Одно из наиболее эффективных средств воздействия на скорость химических реакций - использование катализаторов. Катализаторы - это вещества, которые изменяют скорость реакции, а сами к концу процесса остаются неизменными по составу и по массе. Иначе говоря, в момент самой реакции катализатор активно участвует в химическом процессе, но к концу реакции реагенты изменяют свой химический состав, превращаясь в продукты, а катализатор выделяется в первоначальном виде. Обычно роль катализатора заключается в увеличении скорости реакции, хотя некоторые катализаторы не ускоряют, а замедляют процесс. Явление ускорения химических реакций благодаря присутствию катализаторов носит название катализа, а замедления - ингибирования. Некоторые вещества не обладают каталитическим действием, но их добавки резко увеличивают каталитическую способность катализаторов. Такие вещества называются промоторами. Другие вещества (каталитические яды) уменьшают или даже полностью блокируют действие катализаторов, этот процесс называется отравлением катализатора. Существуют два вида катализа: гомогенный и гетерогенный. При гомогенном катализе реагенты, продукты и катализатор составляют одну фазу (газовую или жидкую). В этом случае отсутствует поверхность раздела между катализатором и реагентами. Особенность гетерогенного катализа состоит в том, что катализаторы (обычно твердые вещества) находятся в ином фазовом состоянии, чем реагенты и продукты реакции. Реакция развивается обычно на поверхности твердого тела. При гомогенном катализе происходит образование промежуточных продуктов между катализатором и реагирующим веществом в результате реакции с меньшим значением энергии активации. При гетерогенномый катализ объясняется адсорбцией реагирующих веществ на поверхности катализатора. В результате этого их концентрация увеличивается и скорость реакции растет. Особым случаем катализа является аутокатализ. Смысл его заключается в том, что химический процесс ускоряется одним из продуктов реакции.