

5.3. Альтерации в синтезе и распаде гема
5.3.1. Порфирии
Синтез гема – многостадийный процесс, в котором участвуют несколько ферментов. В настоящее время описаны наследственные заболевания – порфирии (porphyra – пурпурная краска), причинами которых являются мутации, способные вызвать блок любого из энзимов образования данного продукта (схема 11). В зависимости от органной локализации нарушений генеза порфиринов различают эритропоэтические (в костном мозге) и печёночные (в гепатоцитах). Преобладает аутосомно-доминантный тип наследования.
Подобные недуги встречаются очень редко, несколько чаще диагностируются эритропоэтические порфирии (например, эритропоэтическая протопорфирия вследствие блока феррохелатазы). Из-за неэффективности синтеза гема в клетках эритроидного ряда накапливаются его предшественники, стимулирующие СРО и тем самым провоцирующие гемолиз (развитие гемолитической анемии, желтухи).
В плазме крови повышаются концентрации уропорфириногена, копропорфириногена, которые в нижних отделах мочевыделительной системы окисляются кислородом и в таком виде выделяются с мочой. Обладая сродством к белкам кожи и соединительной ткани, порфирины задерживаются в них, часто стимулируя сдвиги в окраске (красный цвет зубов). Их накопление в эпителии увеличивает чувствительность кожи к УФО, следствием чего могут быть язвы, превращающиеся затем в грубые шрамы. Высок риск их инфицирования с переходом воспаления на хрящевую и костную ткани. Конечности деформируются, возможна тугоподвижность суставов (контрактуры). Для печёночных порфирий более характерны неврологические нарушения (боли по ходу нервных стволов, клиника «острого живота»).
Специфическая диагностика практически отсутствует, базируется на данных анамнеза, клиники. Обращает на себя внимание изменение цвета мочи (часто красно-коричневого) за счёт необычно больших количеств уропорфиринов или копропорфиринов.
Лечение преимущественно симптоматическое. В тяжёлых случаях (при гемолитических кризах) проводят спленэктомию. Необходима защита от солнечных лучей, рекомендуются специальные кремы, мази; днём лучше находиться в затемнённом помещении.
Прогноз обычно неблагоприятный. Смерть провоцируется тяжёлыми кожными поражениями, сниженным иммунитетом, интоксикацией, острыми гемолитическими кризами.

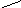 Сукцинил
КоА + Глицин
Сукцинил
КоА + Глицин

 5-аминолевулинатсинтаза
5-аминолевулинатсинтаза
 5-аминолевулиновая
кислота
5-аминолевулиновая
кислота
Дегидратаза 5-АЛК
-
П
 орфирия,
вызванная дефицитом
дегидратазы
орфирия,
вызванная дефицитом
дегидратазы5-АЛК
 Порфобилиноген
Порфобилиноген
М очаПорфобилиногендезаминаза
очаПорфобилиногендезаминаза
уропорфириноген-косинтаза
У ропорфиринI
ропорфиринI
-
В

 рождённая
эритропоэтическая порфирия
рождённая
эритропоэтическая порфирия
У ропорфириногенI
ропорфириногенI
Уропорфириноген III

 Уропорфириноген-
Уропорфириноген-
 декарбоксилаза
декарбоксилаза
-
П
 оздняя
кожная порфирия
оздняя
кожная порфирия
Копропорфириноген I

 Копропорфириноген
III
Копропорфириноген
III
Копропорфириногеноксидаза
 Копропорфирин I
Копропорфирин I
-
Н
 аследственная
копропорфирия
аследственная
копропорфирия
М оча
Протопорфириноген IX
оча
Протопорфириноген IX
Протопорфириногеноксидаза
-
П
 орфирия
variegata
орфирия
variegata
 Протопорфирин
IX
Протопорфирин
IX
 Fe++
Fe++
Феррохелатаза
2Н+
-
Э
 ритропоэтическая
порфирия
ритропоэтическая
порфирия

 Гем
+ Протеин
Гем
+ Протеин

Гемоглобин
Схема 11. Система ферментов синтеза гема в норме и при патологии.