

А. Нематричный синтез полипептидов
![]() Полипептидные
цепи, как известно, являются основой
белков. Полипептидная цепь может быть
представлена обобщенной структурой
(83):
Полипептидные
цепи, как известно, являются основой
белков. Полипептидная цепь может быть
представлена обобщенной структурой
(83):
Концевое звено с группой NH2 называют N-концом, другое концевое звено с группой СООН – С-концом. Полипептиды – частный случай полиамидов, связи CO-NH, соединяющие элементарные звенья полипептидной цепи, называют пептидными связями.
 Мономеры
для синтеза полипептидных цепей -
α-аминокислоты; все они, кроме одной
могут быть представлены формулами
(84)-(84’); одна – пролин – формулами
(85)-(85’):
Мономеры
для синтеза полипептидных цепей -
α-аминокислоты; все они, кроме одной
могут быть представлены формулами
(84)-(84’); одна – пролин – формулами
(85)-(85’):
В средах, близких к нейтральным, аминокислоты существуют почти целиком в форме биполярных ионов (84’) и (85’). Радикалы RI могут быть алифатическими, ароматическими, гетероциклическими, многие из них содержат разнообразные функциональные группы: ОН, NH2, COOH, SH и др. Для обозначения α-аминокислот в литературе используют три буквы (латинские) названия (чаще всего три первые, но не всегда), например Gly (глицин), Val (валин), Trp(триптофан).
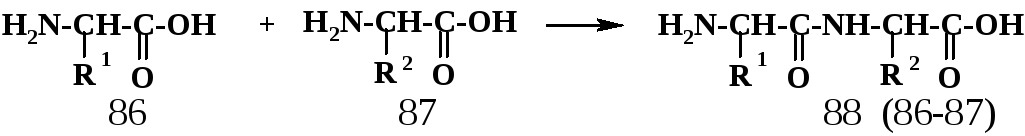 Нематричные
синтезы полипептидных цепей из
α-аминокислот основаны на нескольких
целенаправленных модификациях
функциональных групп; эти модификации
обеспечивают протекание на каждой
стадии единственной
реакции –
взаимодействия карбоксильной функции
предыдущего звена с аминогруппой
последующего (если считать с N-конца).
Необходимость такой модификации можно
проиллюстрировать на простейшем примере
синтеза димера – дипептида, для которого
дан формальный
синтез из мономеров:
Нематричные
синтезы полипептидных цепей из
α-аминокислот основаны на нескольких
целенаправленных модификациях
функциональных групп; эти модификации
обеспечивают протекание на каждой
стадии единственной
реакции –
взаимодействия карбоксильной функции
предыдущего звена с аминогруппой
последующего (если считать с N-конца).
Необходимость такой модификации можно
проиллюстрировать на простейшем примере
синтеза димера – дипептида, для которого
дан формальный
синтез из мономеров:
Для препаративного синтеза дипептида (88) необходимо: А. Защитить группу NH2 аминокислоты (86), чтобы избегнуть вариантов взаимодействия (86)-(86) и (87)-(86); Б. Активировать карбоксильную функцию аминокислоты (86), т.к. сама карбоксильная группа малоактивна в реакциях с нуклеофилами; В. Защитить группу СООН аминокислоты (87); это нужно для того, чтобы эта аминокислота не находилась в виде биполярного иона типа (84’); в такой форме аминогруппа не нуклеофильна и, следовательно, неактивна.
 Поликонденсацию,
ведущую к синтезу пептидной цепи с
заданной первичной структурой, можно
представить следующей схемой:
Поликонденсацию,
ведущую к синтезу пептидной цепи с
заданной первичной структурой, можно
представить следующей схемой:
где Z – защитная группа для аминогруппы; Х – активирующая группа для первой карбоксильной функции; Y – защитная группа для второй карбоксильной функции.
После образования защищенного с двух концов дипептида (89) снимают защитную группу либо с его N-конца (1), либо с его С-конца (2) (совмещая снятие защиты с активированием). Далее освободившуюся группу NH2 в дипептиде (90) или активированную карбоксильную функцию в дипептиде (91) используют для проведения следующей стадии – реакции с очередным модифицированным мономером с образованием трипептида; эта схема повторяется. В варианте (1) пептидная цепь наращивается с С-конца, в варианте (2) - с N-конца. В реакции можно вводить не обязательно модифицированные мономеры, но и «сшивать» пептиды друг с другом.
Приведенная здесь схема упрощенная - реально приходится также защищать некоторые функциональные группы, находящиеся в боковых группах Ri, например, группу NH2 в боковом радикале лизина.
А. Защитные группы. Основные требования к защитным группам: а. Они должны полностью предотвращать участие защищаемой группы в проводимых реакциях (блокировать защищаемую группу); б. После проведения реакции они должны достаточно легко удаляться с регенерацией защищаемой группы и без затрагивания остальных фрагментов продукта реакции (в частности, при синтезе пептидов – без разрыва пептидных связей).
![]() 1.
NH2-Защитные
группы (группы
Z).
Сейчас известно большое число вариантов
эффективной защиты группы NH2;
используются несколько типов защитных
групп. Здесь ограничимся наиболее широко
применяемым типом – уретановыми
защитными группами. Для
их постановки соединение, содержащее
группу NH2,
вводят в реакцию с производным моноэфира
угольной кислоты, например, хлорангидридом
(эфиром хлоругольной кислоты,
хлоркарбонатом):
1.
NH2-Защитные
группы (группы
Z).
Сейчас известно большое число вариантов
эффективной защиты группы NH2;
используются несколько типов защитных
групп. Здесь ограничимся наиболее широко
применяемым типом – уретановыми
защитными группами. Для
их постановки соединение, содержащее
группу NH2,
вводят в реакцию с производным моноэфира
угольной кислоты, например, хлорангидридом
(эфиром хлоругольной кислоты,
хлоркарбонатом):
Кроме хлорангидридов, можно использовать азиды или ангидриды. Группировка RO-CO-NH- называется уретановой, откуда и название защиты. Постановка уретановой защиты – аналог ацилирования аминогруппы; обычное ацилирование производными карбоновых кислот неприменимо, т.к. ацильные защитные группы плохо удаляются; напротив, уретановая защита снимается легко, в мягких условиях, причем в различных, в зависимости от характера радикала R. Приведем три примера:
![]() а.
R=C6H5CH2;
защитная группа называется
бензилоксикарбонильной
(карбобензилокси-защита,
Z-защита);
это исторически первый пример уретановой
защиты группы NH2
(М. Бергман, Л. Зервас, 1932 г.). После
проведения необходимой реакции
бензилоксикарбонильная защита легко
снимается мягким каталитическим
гидрированием (точнее – гидрогенолизом):
а.
R=C6H5CH2;
защитная группа называется
бензилоксикарбонильной
(карбобензилокси-защита,
Z-защита);
это исторически первый пример уретановой
защиты группы NH2
(М. Бергман, Л. Зервас, 1932 г.). После
проведения необходимой реакции
бензилоксикарбонильная защита легко
снимается мягким каталитическим
гидрированием (точнее – гидрогенолизом):
Продукты гидрогенолиза защитной группы – толуол и СО2 – легко удаляются из реакционной среды.
 б.
R
= (CH3)3C;
защитная группа – трет-бутилоксикарбонильная,
Вос-защита (Butyl-
oxycarbonyl);
эта защита легко удаляется при мягкой
кислотной обработке, например, при
действии трифторуксусной кислоты:
б.
R
= (CH3)3C;
защитная группа – трет-бутилоксикарбонильная,
Вос-защита (Butyl-
oxycarbonyl);
эта защита легко удаляется при мягкой
кислотной обработке, например, при
действии трифторуксусной кислоты:
Здесь оба продукта, образующиеся при снятии защиты, газообразны, что еще более облегчает их удаление.
В. R=CH3SO2CH2CH2 – метилсульфонилэтилоксикарбонильная защита (Msc-защита); эта защита снимается NaOH в мягких условиях (рН 10-12, 0 оС).
Различие в условиях снятия приведенных защит позволяет по-разному защищать α-NH2-группу аминокислоты и NH2-группу в боковом радикале лизина. Тогда одну защиту (α-NH2-группы) можно снять, а другую (“лизиновую”) – оставить (защиту боковых групп обычно снимают после окончания формирования полипептидной цепи).
Известно еще несколько вариантов уретановой защиты, а также несколько иных типов защиты группы NH2 - формильная, фталильная, трифторацетильная; сведения об этих способах можно найти в литературе по биоорганической химии.
2. СООН -Защитные группы. Чаще всего используют образование бензиловых или трет-бутиловых эфиров:
Б ензиловые
эфиры обычно получают прямой этерификацией,трет-бутиловые
–присоединением изобутилена при
кислотном катализе (этерификация
трет-бутанолом
пространственно затруднена). Защитные
группы снимаются в мягких условиях,
сходными с условиями снятия соответствующих
уретановых защитных групп.
ензиловые
эфиры обычно получают прямой этерификацией,трет-бутиловые
–присоединением изобутилена при
кислотном катализе (этерификация
трет-бутанолом
пространственно затруднена). Защитные
группы снимаются в мягких условиях,
сходными с условиями снятия соответствующих
уретановых защитных групп.
Иногда для защиты группы СООН используют простое солеобразование:
-СООН → -СОО‾.
![]() Б.
Активирующие группы (группы
Х). Реакции образования пептидной связи
относятся к реакциям ацилирования;
главной стадией таких реакций является
нуклеофильное присоединение (в данном
случае группы NH2)
к связи С=О карбоксильной функции. Как
уже упоминалось, группа СООН довольно
малоактивна в реакциях ацилирования,
т.к. неподеленная пара электронов атома
кислорода группы ОН в значительной
степени компенсирует дефицит электронной
плотности на карбонильном атоме углерода:
Б.
Активирующие группы (группы
Х). Реакции образования пептидной связи
относятся к реакциям ацилирования;
главной стадией таких реакций является
нуклеофильное присоединение (в данном
случае группы NH2)
к связи С=О карбоксильной функции. Как
уже упоминалось, группа СООН довольно
малоактивна в реакциях ацилирования,
т.к. неподеленная пара электронов атома
кислорода группы ОН в значительной
степени компенсирует дефицит электронной
плотности на карбонильном атоме углерода:
Активирующая группа (Х) должна быть электроноакцепторной, чтобы сделать атом углерода карбоксильной группы более электрофильным и облегчить атаку аминогруппы для образования пептидной связи.
Известно достаточно много производных карбоновых кислот, содержащих электроноакцепторные группы, но не все они могут быть использованы; например, непригодна самая очевидная активирующая группа – С1 (т.е. не используются хлорангидриды), т.к. в этом случае не сохраняется конфигурация аминокислоты (происходит рацемизация). Ниже приведены широко используемые варианты активации.
 А.
Образование
активированных эфиров
(Х = OR).
В этом
варианте получают ариловые сложные
эфиры кислот, которые содержат в
ароматическом радикале электроноакцепторные
группы (например, пара-нитрофенильную
или пентафторфенильную):
А.
Образование
активированных эфиров
(Х = OR).
В этом
варианте получают ариловые сложные
эфиры кислот, которые содержат в
ароматическом радикале электроноакцепторные
группы (например, пара-нитрофенильную
или пентафторфенильную):
![]() Б.
Образование
азидов кислот (Х
= N3):
Б.
Образование
азидов кислот (Х
= N3):
Азиды кислот получают через сложные эфиры и гидразиды; азидная группа обладает сильным электроноакцепторным действием
 В.
Образование
смешанных ангидридов. Обычно
используют смешанные эфиры
α-аминокислот
и производных угольной (92) или фосфорной
(93) кислот:
В.
Образование
смешанных ангидридов. Обычно
используют смешанные эфиры
α-аминокислот
и производных угольной (92) или фосфорной
(93) кислот:
Получение смешанных ангидридов с производными угольной кислоты удобно тем, что при последующем образовании пептидной связи активирующая группа удаляется в виде спирта и СО2, что препаративно удобно:
О![]() бразование
смешанных ангидридов α-аминокислот с
производным фосфорной кислоты
(аминоациладенилатов) – важная реакция,
предшествующая процессу биосинтеза
белков - трансляции.
бразование
смешанных ангидридов α-аминокислот с
производным фосфорной кислоты
(аминоациладенилатов) – важная реакция,
предшествующая процессу биосинтеза
белков - трансляции.
Г. Использование карбодиимидов Применение карбодиимидов R-N=C=N-R1 позволяет провести активацию карбоксильной группы и образование пептидной связи в одну стадию, без выделения активированной аминокислоты (или пептида). Если, допустим, прибавить карбодиимид к смеси NH2-защищенной первой аминокислоты и СООН-защищенной второй аминокислоты, то протекают две последовательные реакции:
В начале
карбодиимид реагирует с карбоксильной
группой первой аминокислоты с образованием
ее активированного производного (94)
(напоминающего смешанный ангидрид);
далее это производное реагирует с
группойNH2
второй аминокислоты, причем образуется
пептид, а активирующая группа удаляется
в виде симм.
дизамещенной мочевины.
начале
карбодиимид реагирует с карбоксильной
группой первой аминокислоты с образованием
ее активированного производного (94)
(напоминающего смешанный ангидрид);
далее это производное реагирует с
группойNH2
второй аминокислоты, причем образуется
пептид, а активирующая группа удаляется
в виде симм.
дизамещенной мочевины.
Одним из наиболее широко применяемых реагентов этого типа является дициклогексилкарбодиимид (DCC) (R = R1 =циклогексил); в ходе пептидного синтеза из него образуется симм. дициклогексилмочевина, нерастворимая в большинстве органических растворителей и легко отделяемая фильтрованием. Также широко используются водорастворимые карбодиимиды [например, R = Et, R1 = (CH2)3N(CH3)2].
Карбодиимиды используются не только в пептидном синтезе, но и при синтезе in vitro полинуклеотидов (см. ниже).
 Д.
Использование
N-карбоксиангидридов.
Этот вариант
позволяет совместить
защиту
аминогруппы и активацию карбоксильной
функции. N-Карбоксиангидриды
(ангидриды Лейхса) образуются при
взаимодействии α-аминокислот с фосгеном:
Д.
Использование
N-карбоксиангидридов.
Этот вариант
позволяет совместить
защиту
аминогруппы и активацию карбоксильной
функции. N-Карбоксиангидриды
(ангидриды Лейхса) образуются при
взаимодействии α-аминокислот с фосгеном:
П ри
этом совмещаетсязащита
группы NH2
по уретановому типу и активация
карбоксильной
группы по типу образования смешанного
ангидрида с производным угольной
кислоты. Образование полипептидов при
использовании N-карбоксиангидридов
идет следующим образом:
ри
этом совмещаетсязащита
группы NH2
по уретановому типу и активация
карбоксильной
группы по типу образования смешанного
ангидрида с производным угольной
кислоты. Образование полипептидов при
использовании N-карбоксиангидридов
идет следующим образом:
Взаимодействие N-карбоксиангидрида с солью второй аминокислоты при точно установленном значении рН 10,2 приводит к образованию пептидной связи и получению соли производного дипептида (95), содержащей фрагмент соли карбаминовой кислоты. При слабом подкислении (рН 5) образующийся фрагмент карбаминовой кислоты немедленно декарбоксилируется (производные карбаминовой кислоты со свободной группой СООН весьма легко декарбоксилируются), т.е. происходит снятие защиты с N-конца дипептида. Далее полученный дипептид (96) вводят в реакцию с очередным N-карбоксиангидридом при рН 10,2 и т.д.
 Этот
вариант, в принципе, позволяет сократить
число стадий пептидного синтеза, но он
требует точного
соблюдения
условий, в частности, поддержания точного
значения рН. В других условиях может
произойти, в частности, образование
гомополимеров
гомополипептидов
из
N-карбоксиангидридов
по схеме:
Этот
вариант, в принципе, позволяет сократить
число стадий пептидного синтеза, но он
требует точного
соблюдения
условий, в частности, поддержания точного
значения рН. В других условиях может
произойти, в частности, образование
гомополимеров
гомополипептидов
из
N-карбоксиангидридов
по схеме:
Такие гомополипептиды могут служить моделями (хотя и довольно приближенными) природных полипептидов, поэтому их получение имело практическое применение.
Пептидный синтез на полимерных носителях. Как видно из изложенного выше, синтез полипептидных цепей сколько-нибудь значительной длины включает большое число отдельно проводимых стадий десятки, а то и сотни). Это весьма трудоёмкий процесс; кроме того, требуется высочайшая эффективность каждой стадии, сведение к минимуму потерь образующихся пептидов. Эффективность во многом определяется сравнительной растворимостью пептидов и других продуктов реакций, которые нужно отделить от пептида: если растворимость разная, разделение и очистка упрощаются.
Методика пептидного синтеза на полимерном носителе значительно упрощает процедуру синтеза и, в частности, кардинально решает проблему растворимости, что позволяет повысить эффективность синтеза. Идея синтеза состоит в том, что формируемая полипептидная цепь с самого начала синтеза связана с макромолекулой полимера-носителя и лишь в конце синтеза отделяется от нее.
Наибольшее распространение имеет использование нерастворимого полимера-носителя (твердофазный пептидный синтез); эта методика впервые была предложена Р. Меррифилдом в 1963 г. В качестве полимера-носителя обычно используется частично хлорметилированный сополимер стирола с небольшим количеством 1,4-дивинилбензола; это пространственный полимер с редкими поперечными сшивками между цепями и определенным количеством групп СН2С1:
П ептидный
синтез на носителе протекает по схеме:
ептидный
синтез на носителе протекает по схеме:
 Вначале
первую аминокислоту (NH2-защищенную,
чаще всего Вос-защитой) «прикрепляют»
к полимеру-носителю за счет взаимодействия
хлорметильной группы с карбоксильной
группой аминокислоты (точнее карбоксилатной,
в которую она превращается в присутствии
триэтиламина); аминокислота прикрепляется
к полимеру, образуя с ним сложный эфир
типа бензилового (97). Далее снимают
защиту с группы NH2,
добавляют вторую NH2-защищенную
аминокислоту (обычно в присутствии
карбодиимида); образуется прикрепленный
к полимеру N-защищенный
дипептид (98). Далее цикл повторяют:
снимают защиту Z,
добавляют третью аминокислоту и т.д.;
происходит наращивание пептидной цепи
с С-конца по схеме линейного синтеза.
Вначале
первую аминокислоту (NH2-защищенную,
чаще всего Вос-защитой) «прикрепляют»
к полимеру-носителю за счет взаимодействия
хлорметильной группы с карбоксильной
группой аминокислоты (точнее карбоксилатной,
в которую она превращается в присутствии
триэтиламина); аминокислота прикрепляется
к полимеру, образуя с ним сложный эфир
типа бензилового (97). Далее снимают
защиту с группы NH2,
добавляют вторую NH2-защищенную
аминокислоту (обычно в присутствии
карбодиимида); образуется прикрепленный
к полимеру N-защищенный
дипептид (98). Далее цикл повторяют:
снимают защиту Z,
добавляют третью аминокислоту и т.д.;
происходит наращивание пептидной цепи
с С-конца по схеме линейного синтеза.
Растущая пептидная цепь с самого начала (с первого звена) нерастворима, т.к. ковалентно связана с пространственным полимером, который по определению нерастворим [в то же время пространственная сетка редкая; поэтому полимер может набухать в раст-ворителе, и реагенты имеют свободный доступ к N-концу растущей цепи]. Поэтому все побочные продукты (прежде всего избыток реагента) легко удаляются промывкой, экстракцией или фильтрованием полимера [реагенты на каждой стадии берут в большом избытке, чтобы обеспечить полноту протекания каждой реакции]. Это существенно повышает эффективность синтеза.
 По
окончании формирования требуемой
пептидной цепи ее отсоединяют от
полимера-носителя (например, действием
смеси HBr-CF3COOH
в мягких условиях); одновременно снимается
защита с N-конца
(если это Вос-защита):
По
окончании формирования требуемой
пептидной цепи ее отсоединяют от
полимера-носителя (например, действием
смеси HBr-CF3COOH
в мягких условиях); одновременно снимается
защита с N-конца
(если это Вос-защита):
Твердофазный синтез пептидов автоматизирован и осуществляется на специальных устройствах – синтезаторах. Наибольшие успехи достигнуты при синтезе олигопептидов (порядка 8-15 звеньев); однако этим способом можно получать и высокомолекулярные полипептиды; в частности, одним из первых значительных достижений твердофазного синтеза был синтез фермента рибонуклеазы, содержащей 124 звена.
Одной из проблем, с которой сталкивается твердофазный синтез, является уменьшение степени набухания полимера по мере роста пептидной цепи; это затрудняет доступ к группам NH2 растущей полимерной цепи. В этом случае реакция постановки очередного звена может пройти неполностью, частично образуется пептид с «пропуском» звена, который, как правило, уже не обладает нужной биологической активностью (пропуск хотя бы одного звена в полипептидной цепи меняет ее пространственную организацию, а, следовательно, и биологическую активность). Поэтому такие «ложные» пептиды необходимо отделять от «правильных», что достаточно трудно.
Проблема, по крайней мере, частично, решается при использовании в качестве носителей растворимых полимеров; в качестве таких носителей можно использовать линейные полимеры – полистирол, полиэтиленгликоли или полиуретаны. В этом варианте синтез ведется в растворе, где доступ реагентов к растущей цепи облегчен по сравнению с твердофазным синтезом. Затем полимер с «привязанной» к нему растущей пептидной цепью осаждают «плохим» растворителем, отфильтровывают от остальных продуктов, опять растворяют в «хорошем» растворителе и продолжают синтез. Этот вариант, предложенный М. М. Шемякиным, называют жидкофазным пептидным синтезом; он используется для синтеза олигопептидов; при синтезе высокомолекулярных полипептидов меняется растворимость полимера, что создает ряд проблем.
Нематричный лабораторный синтез пептидов (во всех вариантах) используется в настоящее время преимущественно для синтеза природных олигопептидов; синтез природных белков более эффективно осуществляется биотехнологически – путем встраивания генов, кодирующих белки, в рекомбинантные ДНК, с последующими клонированием и экспрессией этих генов.