

Общие закономерности поликонденсации
Ступенчатый характер полимеризации. Все реакции поликонденсации протекают по ступенчатому механизму, который уже рассматривался в разделе «Ступенчатая полимеризация». Формирование макромолекулы протекает путем повторяющихся реакций конденсации, причем на любой стадии формирования образуются обычные молекулы, не содержащие активных центров. При этом молекулы любой величины содержит активные функциональные группы (при линейной поликонденсации – концевые, при трехмерной – также и не концевые) и сохраняют возможность дальнейшего вступления в реакции конденсации с увеличением макромолекулы.
Формирование цепи при линейнойполиконденсации может протекать по трем схемам:
1. В реакцию конденсации могут вступать присутствующие в реакционной смеси молекулы любой степени полимеризации, т.е. в общем случае реагируют m-мер и n-мер:
a-[-R-]m-b + a-[-R-]n-b → a-[R-]m+n-b + ab
m, n =1, 2, 3…
Естественно, в результате такой единичной реакции цепь увеличивается на различное количество звеньев. По этой схеме протекает формирование макромолекулярных цепей практически всех синтетических полимеров при обычной (непрограммируемой) поликонденсации.
Для программируемой поликонденсации, в частности, при матричном синтезе нуклеиновых кислот и белков реализуются две другие схемы:
2. В каждой реакции конденсации участвует растущая цепь и молекула мономера; в ре-
зультате единичной реакции цепь увеличивается на одно звено. Иногда такую схему называют «линейной». Чисто формально эта схема напоминает рост цепи при цепной полимеризации, однако это не цепной, а типично ступенчатый механизм. По «линейной» схеме протекает биосинтез многих биополимеров. Один пример – синтез натурального каучука и гуттаперчи – был приведен выше (стр. 63). По этой же схеме проходят матричные синтезы нуклеиновых кислот (репликация на «ведущей» цепи ДНК и транскрипция) и белков (трансляция). Из чисто синтетических реакций по этой схеме происходит ступенчатая полимеризация циклических мономеров (стр. 50).
3. Известны примеры «гибридной» схемы, когда отдельные участки полимерной цепи синтезируются по «линейной» схеме, а затем эти участки «сшиваются». Такую схему иногда называют «конвергентной» или «блочной» . По такой схеме идет репликация на одной из двух цепей ДНК – «отстающей» (синтез и сшивание фрагментов Оказаки). По этой же схеме в ряде случаев проводят лабораторный (программируемый) синтез полипептидов и полинуклеотидов.
Рассмотрим обычную непрограммируемую поликонденсацию (при которой формирование цепи идет по варианту 1). Вначале рассмотрим как меняется реакционная система по мере увеличения глубины процесса.
Уже сама схема формирования полимерных цепей приводит к выводу, что характер изменения реакционной системы в ходе реакции здесь резко отличается от случая цепной полимеризации:
А. Если при цепной полимеризации сразу образуются большие макромолекулы, то при поликонденсации рост макромолекул происходит постепенно; скорость формирования макромолекул здесь на несколько порядков ниже; молекулярная масса продукта поликонденсации растет по мере увеличения глубины процесса.
Б. Если при цепной полимеризации количество мономера постепенно уменьшается по мере протекания процесса, то при поликонденсации основная масса мономера исчезает уже при малых глубинах процесса: его молекулы объединяются вначале в олигомерные продукты (димеры, тримеры и т. д.), а затем происходит дальнейшее укрупнение молекулы за счет взаимодействия их между собой (а также с оставшимися молекулами мономера). Чисто механическая аналогия такого хода поликонденсации – образование сначала очень маленьких капелек жидкости, а затем постепенное их слияние во все более крупные капли. Поэтому при поликонденсации под глубиной процесса понимают не степень конверсии мономера (как при цепной полимеризации), а долю (или процент) прореагировавших функциональных групп.
В. Если при цепной полимеризации «мертвый» полимер обычно «выключается» из процесса синтеза [за исключением «оживающих» полимеров, полученных при инициировании инифертерами (cтр. 27-28)], то при поликонденсации любая молекула независимо от ее величины может реагировать дальше, т.к. содержит активные функциональные группы.
Может показаться, что логическим финалом поликонденсации должно быть сшивание всех цепей в одну гигантскую «супермолекулу». В случае трехмерной поликонденсации так обычно и происходит; в случае линейной поликонденсации этого не происходит никогда. Действительно, для сшивания всех цепей при линейной поликонденсации необходимо, чтобы прореагировали все функциональные группы (кроме двух, которые стали бы концевыми в этой супермолекуле). Этому препятствует целый ряд факторов, наиболее важные из которых рассмотрены ниже.
Далее рассмотрим основные факторы, определяющие ход и результаты непрограммированной поликонденсации.
1. Влияние соотношения реагирующих групп при линейной поликонденсации на молекулярную массу полимера. При линейной поликонденсации для достижения максимально возможной молекулярной массы соотношение реагирующих функциональных групп должно быть строго эквивалентным. Если же одна из групп находится в недостатке, она будет исчерпана, глубина процесса относительно этой группы станет равной единице (100%), и рост полимерных цепей прекратится.
Допустим, в поликонденсацию вступают мономеры a-R1-a и b-R2-b, причем на n+1 молекул первого мономера приходится n молекул второго. Поликонденсация протекает следующим образом (представлен среднестатистический результат):
В начале
реагируютn
молекул первого мономера и n
молекул второго с образованием
n-мера
(70), а затем избыточная молекула первого
мономера блокирует «b-конец»
n-мера
с образованием полимера (71) с одинаковыми
группами (а) на обоих концах молекулы.
Поскольку
одинаковые группы друг с другом в
условиях реакции не реагируют,
поликонденсация прекращается вследствие
исчерпания группы b;
средняя степень полимеризации не может
превысить величины n.
Легко убедиться, что уже 1%-й молярный
избыток одного из мономеров не позволяет
получить среднюю степень полимеризации
выше 100.
начале
реагируютn
молекул первого мономера и n
молекул второго с образованием
n-мера
(70), а затем избыточная молекула первого
мономера блокирует «b-конец»
n-мера
с образованием полимера (71) с одинаковыми
группами (а) на обоих концах молекулы.
Поскольку
одинаковые группы друг с другом в
условиях реакции не реагируют,
поликонденсация прекращается вследствие
исчерпания группы b;
средняя степень полимеризации не может
превысить величины n.
Легко убедиться, что уже 1%-й молярный
избыток одного из мономеров не позволяет
получить среднюю степень полимеризации
выше 100.
![]() К
тем же последствиям (исчерпанию одной
из групп) приводит наличие примеси
монофункционального
соединения,
R3-a
или R3-b,
содержащего группу того же типа, что и
мономеры:
К
тем же последствиям (исчерпанию одной
из групп) приводит наличие примеси
монофункционального
соединения,
R3-a
или R3-b,
содержащего группу того же типа, что и
мономеры:
Монофункциональные примеси ограничивают величину цепи и при поликонденсации мономеров типа a-R-b, которые сами по себе содержат реагирующие группы в эквивалентном количестве; эти примеси нарушают эквивалентность.
Практические выводы, вытекающие из фактора соотношения реагирующих групп:
А. Если необходимо получить полимер с высокой молекулярной массой, то следует:
а). Избегать монофункциональных примесей и вообще использовать максимально чистые мономеры. Если в поликонденсацию вводят два мономера, необходима точная дозировка. В качестве иллюстрации к сказанному можно привести поликонденсацию адипиновой кислоты и гексаметилендиамина, приводящую к полиамиду, из которого получают волокно «найлон 6,6». Перед проведением поликонденсации компоненты смешивают в мягких условиях; при этом образуется соль (так называемая АГ-соль). Эта соль легко очищается перекристаллизацией; в ней соотношение реагирующих групп эквивалентно. При нагревании этой соли до достаточно высокой температуры происходит поликонденсация:
б
![]() )
При проведении поликонденсации необходимо
избегать протекания побочных реакций,
которые могли бы нарушить эквивалентность
реагирующих групп. Простой пример –
декарбоксилирование дикарбоновых
кислот при нагревании до высоких
температур:
)
При проведении поликонденсации необходимо
избегать протекания побочных реакций,
которые могли бы нарушить эквивалентность
реагирующих групп. Простой пример –
декарбоксилирование дикарбоновых
кислот при нагревании до высоких
температур:
Поэтому, в частности, упомянутую выше поликонденсацию АГ-соли необходимо проводить при строгом контроле температуры и в атмосфере инертного газа.
Б. Если необходимо получить продукты с ограниченной молекулярной массой, в частности, олигомеры, то фактор эквивалентности используют для регулирования молекулярной массы: создают рассчитанный избыток одной из функциональных групп (дозировкой при поликонденсации двух мономеров или добавкой монофункциональной примеси при гомополиконденсации).
2. Влияние обратимости поликонденсации на молекулярную массу полимера. Если соотношение реагирующих функциональных групп строго эквивалентно, то размеры линейных полимерных цепей ограничивает состояние равновесия реакций поликонденсации. Состояние равновесия определяет максимально возможную долю прореагировавших групп, т.е. максимально достижимую глубину поликонденсации. Эта глубина не может достигать значения, равного единице (100%), потому что для этого должны прореагировать все функциональные группы, а это невозможно из-за обратимости любой реакции поликонденсации, т.е. протекания обратной реакции – деструкции полимера низкомолекулярным продуктом, образовавшимся в ходе процесса.
В зависимости от природы мономеров, степень обратимости поликонденсации может меняться в очень широких пределах. Принято разделение реакций поликонденсации на две группы: равновесную и неравновесную. Для равновесной поликонденсации обратимость выражена в заметной степени, для неравновесной -–лишь в незначительной степени. Границей здесь считается величина константы равновесия, равная 103; процессы равновесной поликонденсации имеют меньшие значения констант (обычно они значительно меньшие); процессы неравновесной поликонденсации – бóльшие (чаще всего – значительно).
К реакциям равновесной поликонденсации относятся, прежде всего, чрезвычайно широко применяемые на практике варианты реакций полиацилирования – взаимодействие дикарбоновых кислот и их эфиров с двухатомными спиртами (фенолами) и диаминами – в настоящее время это одни из основных промышленных способов получения полиэфиров и полиамидов Процессы биосинтеза белков и нуклеиновых кислот также относятся к реакциям равновесной поликонденсации. Из других типов реакций к этой группе относятся поликоординация, образование полиацеталей из карбонильных соединений и спиртов.
Из реакций неравновесной поликонденсации прежде всего следует отметить другие варианты реакций полиацилирования - взаимодействие дихлорангидридов дикарбоновых кислот с двухатомными спиртами (фенолами или фенолятами) и диаминами. К этой же группе реакций относятся окислительная дегидрополиконденсация, ряд реакций нуклеофильного замещения.
 Проведение
реакций равновесной и неравновесной
поликонденсации требует различных
подходов. Это становится ясным из
зависимости средней
степени полимеризации
продукта реакции от состояния
равновесия.
Оно выражается простым уравнением:
Проведение
реакций равновесной и неравновесной
поликонденсации требует различных
подходов. Это становится ясным из
зависимости средней
степени полимеризации
продукта реакции от состояния
равновесия.
Оно выражается простым уравнением:
где K – константа равновесия соответствующей реакции конденсации;
[ab] – концентрация низкомолекулярного продукта, выделившегося при поликонденсации
Из уравнения ясно, что при равновесной поликонденсации, когда значение К невелико, для достижения приемлемых значений Р (соответствующих молекулярным массам порядка 20-50 тысяч) необходимо удалять из сферы реакции низкомолекулярный продукт. Расчет показывает, что, например, при полиэтерификации (K~ 10) для получения полиэфира с Р = 100 (М порядка 25000) необходимо удалять выделяющуюся воду до остаточной концентрации порядка 0,001%. Поэтому реакции равновесной конденсации проводят в таких условиях, когда низкомолекулярный продукт удаляется (например, отгонкой, в конце процесса – в вакууме). Удаление побочного продукта поликонденсации осуществляется и при биосинтезе: пирофосфат, выделяющийся при матричном синтезе ДНК или РНК (см. стр. 61), немедленно гидролизуется до фосфата; при матричном синтезе белка удаляется освободившаяся в ходе синтеза транспортная РНК; это позволяет получать весьма высокомолекулярные биополимеры.
Для неравновесной поликонденсации (большие величины К) удалять низкомолекулярный продукт необязательно, хотя иногда и здесь пользуются этим приемом.
Далее объединим влияние двух этих факторов – соотношения реагирующих групп и состояния равновесия - и приведем (без вывода) уравнение зависимости средней степени полимеризации от глубины процесса и соотношения реагирующих групп (максимально достижимая глубина процесса связана с состоянием равновесия, см. выше):
г![]() деr
– стехиометрический
баланс –
отношение числа функциональных групп,
находящихся в недостатке,
к числу избыточных групп; при эквивалентности
групп r
= 1, при неэквивалентности r
< 1.
деr
– стехиометрический
баланс –
отношение числа функциональных групп,
находящихся в недостатке,
к числу избыточных групп; при эквивалентности
групп r
= 1, при неэквивалентности r
< 1.
р – глубина процесса (т.е. доля прореагировавших групп, находящихся в недостатке).
Уравнение позволяет прогнозировать и планировать среднюю молекулярную массу полимера при поликонденсации. Рассмотрим некоторые частные случаи:
![]() 1.
Пусть р = 1, т.е. прореагировали все
функциональные группы, находящиеся в
недостатке (при
эквивалентном соотношении реагирующих
групп глубина не может достичь единицы
из-за обратимости реакции). При этом
уравнение приобретает вид:
1.
Пусть р = 1, т.е. прореагировали все
функциональные группы, находящиеся в
недостатке (при
эквивалентном соотношении реагирующих
групп глубина не может достичь единицы
из-за обратимости реакции). При этом
уравнение приобретает вид:
Например, если r = 0,95, то P = 1,95 : 0,05 = 39 (т.е. полимер с небольшой молекулярной массой).
![]() 2.
Пусть соотношение реагирующих групп
строго
эквивалентно
(r
= 1). В этом случае:
2.
Пусть соотношение реагирующих групп
строго
эквивалентно
(r
= 1). В этом случае:
Это уравнение – частный случай уравнения Карозерса (см. ниже) для линейной поликонденсации (уравнение Карозерса охватывает также и трехмерную поликонденсацию). Из уравнения, например, следует, что если Р = 100, то р=0,99, т.е. для достижения более или менее высокомолекулярного соединения необходимо провести реакцию до весьма большой глубины процесса – а это возможно только при равновесии, сильно сдвинутом вправо.
![]() Все
сказанное выше относилось к линейной
поликонденсации.
При трехмерной
поликонденсации
ограничения, препятствующие увеличению
молекулярной массы, гораздо менее
строгие. В общем случае (и для линейной
и для трехмерной поликонденсации)
средняя степень полимеризации определяется
по уравнению
Карозерса:
Все
сказанное выше относилось к линейной
поликонденсации.
При трехмерной
поликонденсации
ограничения, препятствующие увеличению
молекулярной массы, гораздо менее
строгие. В общем случае (и для линейной
и для трехмерной поликонденсации)
средняя степень полимеризации определяется
по уравнению
Карозерса:
где р – глубина процесса; f – средняя функциональность мономеров, участвующих в реакции. Величина f подсчитывается следующим образом:
 А.
Если соотношение реагирующих групп
эквивалентно,
то:
А.
Если соотношение реагирующих групп
эквивалентно,
то:
где Ni – относительное число молекул мономера, имеющего функциональность fi.
Н апример,
при поликонденсации трехосновной
кислоты и двухатомного спирта приэквивалентном
соотношении
реагирующих групп кислота и спирт должны
реагировать в молярном соотношении
2:3:
апример,
при поликонденсации трехосновной
кислоты и двухатомного спирта приэквивалентном
соотношении
реагирующих групп кислота и спирт должны
реагировать в молярном соотношении
2:3:
Б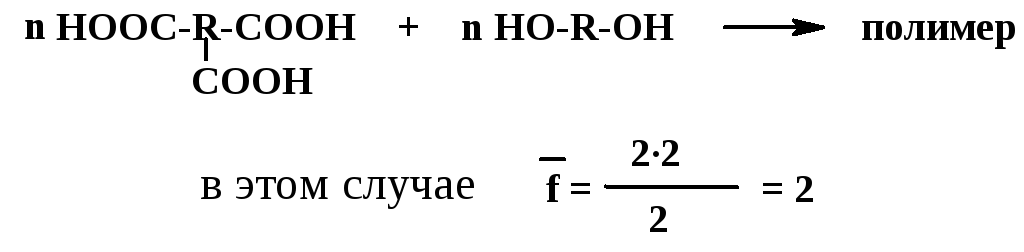 .
Если соотношение реагирующих групп
неэквивалентное, то средняя функциональностьf
определяется как отношение удвоенного
числа групп, находящихся в
недостатке, к
общему числу молекул. Например, при
поликонденсации тех же мономеров, что
и в случае А, но в молярном соотношении
1:1, в недостатке будут группы ОН
.
Если соотношение реагирующих групп
неэквивалентное, то средняя функциональностьf
определяется как отношение удвоенного
числа групп, находящихся в
недостатке, к
общему числу молекул. Например, при
поликонденсации тех же мономеров, что
и в случае А, но в молярном соотношении
1:1, в недостатке будут группы ОН
Из уравнения Карозерса легко подсчитать ту минимальную глубину поликонденсации, при которой произойдет сшивание всех цепей в единую «супермолекулу». В сшитой структуре Р → ∞, т.е. 2 – рf =0 и р = 2 / f. В приведенном выше примере, где f = 2,4 сшивание происходит при минимальной глубине процесса 2 / 2,4 = 0,83; таким образом, в этом примере сшивание происходит еще достаточно далеко от полного завершения реакции.
При линейной поликонденсации f = 2 и уравнение Карозерса приобретает вид, приведенный выше для линейной поликонденсации с эквивалентным соотношением групп.
3. Зависимость молекулярно-массового распределения при линейной поликонденсации от глубины процесса.
Для линейной поликонденсации (прежде всего равновесной) можно рассчитать не только среднюю степень полимеризации, но и молекулярно-массовое распределение (полидисперсность). Это было впервые сделано одним из основателей современных представлений о поликонденсации П. Флори; соответствующие уравнения называют уравнениями распределения по Флори. В основе расчета лежит сформулированное Флори положение о том, что реакционная способность функциональных групп не зависит от размеров реагирующих молекул (т.е. она одинакова как в мономере, так и в продукте реакции с любым числом элементарных звеньев). Из этого положения следует, что вероятность вступления (или не вступления) в реакцию любой функциональной группы одинакова.
Исходя из этого, можно рассчитать вероятность образования продукта поликонденсации с любым числом звеньев. Рассчитаем вероятность образования х-мера, т.е. продукта поликонденсации, содержащего х мономерных звеньев.
Для образования х-мера необходимо, чтобы произошло х-1 взаимодействие между функциональными группами (например, для образования димера – одно взаимодействие). В то же время, для того, чтобы величина молекулы ограничилась х-мером, необходимо, чтобы одно взаимодействие (под номером х) не произошло. Вероятность протекания одного взаимодействия равна глубине прохождения процесса р (действительно, если реакция не идет и р = 0, вероятность любого взаимодействия равна нулю, а при гипотетическом р = 1, когда все группы прореагировали, вероятность любого взаимодействия равна единице). Вероятность непротекания одного взаимодействия, очевидно, равна 1-р. Поскольку вероятность последовательного протекания нескольких независимых событий равна произведению вероятностей протекания каждого из них, вероятность образования х мера (х-1 взаимодействия и одного не взаимодействия) определяется как:
Wx-мер = рх-1(1-р)
Очевидно, Wx-мер соответствует численной доле молекул х-мера по отношению к общему числу молекул в системе, т.е. Nx
― = рх-1(1-р)
N
Где Nx – число молекул х-мера, N – общее число молекул в системе.
Это выражение называют численным молекулярно-массовым распределением по Флори или уравнением Флори для численного распределения. Поскольку глубина процесса р < 1, из этого уравнения следует, что чем меньше молекула, тем больше ее численная доля. Особенно отчетливо это выражено, при неполной завершенности реакции, когда величина р заметно меньше единицы. По мере большей степени завершенности реакции, когда р приближается к единице, распределение становится более равномерным, но закономерность сохраняется. Зависимость числа молекул х-мера от величины х отражена на рис. 7:
Р ис.7.
Зависимость числа молекул в системе от
их величины при разных глубинах
поликонденсации (р1
< p2
< p3).
ис.7.
Зависимость числа молекул в системе от
их величины при разных глубинах
поликонденсации (р1
< p2
< p3).
Из графика видно, что даже при больших глубинах процесса количество малых молекул относительно велико, а в наибольшем количестве, очевидно, находятся молекулы мономера. Это, на первый взгляд, противоречит сделанному ранее утверждению о том, что при поликонденсации основная масса мономера исчезает при малых глубинах процесса (стр. 70). Противоречия здесь, однако, нет, т.к. там речь идет об исчезновении основной массы мономера, т.е. его массовой доли, а не численной. Массовое распределение молекул в системе, в зависимости от их размеров, дает совсем иную картину, нежели численное.
Для вывода массового распределения (массовой доли х-мера) необходимо учесть число звеньев х в молекуле х-мера. Если в исходной системе (до начала поликонденсации) было N0 молекул мономера, то массовая доля х-мера (Mx) выражается как:
x · Nx
Mx = ――
N0
Общее число молекул после протекания поликонденсации (N) можно выразить через число молекул мономера (N0) как N = N0(1-p) (действительно, чем больше глубина процесса, тем меньше общее число молекул в в системе). Если теперь подставить это выражение в уравнение численного распределения по Флори (см. выше) и провести ряд преобразований, то получим:
x · Nx
―― = х(1-р)2рх-1 , т.е. Мх = х(1-р)2рх-1
N0
Последнее выражение – уравнение массового распределения по Флори. Легко заметить, что весовое распределение существенно отличается от численного, т.к. в правую часть уравнения входит величина х. График массового распределения для разных глубин поликонденсации представлен на рис. 8:
Р ис.8.
Зависимость массовой доли молекул в
системе от их величины при разных
глубинах поликонденсации (р1
< p2
< p3).
ис.8.
Зависимость массовой доли молекул в
системе от их величины при разных
глубинах поликонденсации (р1
< p2
< p3).
Здесь зависимость имеет вид кривой с максимумом, который смещается в сторону более высоких масс при увеличении глубины процесса; максимальную массовую долю имеет х-мер, для которого : 1+ р
х = ———
1 – р
Из графика видно, что массовая доля мономера мала уже при небольших глубинах поликонденсации.
Распределения по Флори наиболее точно соблюдаются при равновесной поликонденсации; немалую роль в этом играют обменные реакции, которые будут рассмотрены ниже.
4. Влияние температуры на ход и результат поликонденсации.
Повышение температуры, естественно, увеличивает скорость поликонденсации. В ряде случаев, когда реагирующие группы довольно малоактивны, необходимо увеличивать температуру (иногда выше 200 оС). Типичный пример – поликонденсация с участием карбоновых кислот или их эфиров. Если реагирующие группы активны (например, хлорангидриды карбоновых кислот), реакции протекают быстро уже при обычных температурах. [Сказанное не относится к программируемой поликонденсации с ферментативным катализом: синтез белка при трансляции, где реагируют эфиры аминокислот (с т-РНК), в обычных условиях проходят с такими скоростями, что пептидная цепь из 200 звеньев образуется за несколько секунд].
Реакции поликонденсации в большинстве случаев слабо экзотермичны, так что повышении температуры равновесие несколько смещается в неблагоприятную сторону. Особого значения это не имеет, т.к. при повышенных температурах проводят обычно равновесную поликонденсацию, где равновесие смещают в нужную сторону удалением низкомолекулярного продукта.
Стоит еще раз отметить, что при повышении температуры возрастает вероятность побочных реакций (стр. 71); поэтому высокотемпературные реакции часто проводят в атмосфере инертного газа, чтобы понизить вероятность побочных процессов.
5. Влияние концентрации мономеров. Реакции конденсации, лежащие в основе поликонденсационных процессов, как правило, бимолекулярны, поэтому скорость поликонденсации имеет второй порядок по мономеру. Если реагирующие функциональные группы относительно малоактивны, этот фактор следует учитывать, и реакции обычно ведут без растворителя. Если реагирующие группы активны, поликонденсацию можно вести и в растворе.
Особый аспект возникает, если с реакциями поликонденсации конкурируют реакции внутримолекулярной циклизации (см. ниже). В этих случаях уменьшение концентрации мономера благоприятствует внутримолекулярным реакциям в ущерб межмолекулярным и, следовательно, не способствует поликонденсации.
6. Влияние катализатора. Ряд реакций поликонденсации проводят с участием катализаторов, например, солей меди (окислительная дегидрополиконденсация), кислот и оснований (реакции получения фенол-формальдегидных и мочевино-формальдегидных полимеров, иногда – реакции полиэтерификации). Катализаторы, естественно, не влияют на состояние равновесия, но позволяют понизить температуру и избежать высокотемпературных побочных процессов. Однако в некоторых случаях катализаторы сами стимулируют побочные процессы; такие варианты необходимо учитывать и стараться избегать.
7. Влияние деструктивных обменных реакций. В ходе процессов поликонденсации, в первую очередь, равновесной, помимо обратной реакции - деструкции полимера низкомолекулярным продуктом поликонденсации – протекают так называемые деструктивные обменные реакции, также сопровождающиеся расщеплением макромолекул ВМС. Особенно характерны такие реакции для равновесных вариантов полиацилирования. Чаще всего деструкция молекулы полимера идет под действием концевой функциональной группы соединения, участвующего в процессе поликонденсации.
А. Алкоголиз при синтезе полиэфиров:
Э то
– один из вариантов обычной реакции
переэтерификации. Деструктирующий
реагент (72) может иметь любую величину
– быть мономером, олигомером или
полимером; в результате реакции
расщепляется сложноэфирная связь и
вытесняется соединение (73) с концевой
группой ОН; если деструктируемая молекула
достаточно большая, то соединение (73)
по теории вероятности, скорее всего,
будет полимером.
то
– один из вариантов обычной реакции
переэтерификации. Деструктирующий
реагент (72) может иметь любую величину
– быть мономером, олигомером или
полимером; в результате реакции
расщепляется сложноэфирная связь и
вытесняется соединение (73) с концевой
группой ОН; если деструктируемая молекула
достаточно большая, то соединение (73)
по теории вероятности, скорее всего,
будет полимером.
Б. Аминолиз при образовании полиамидов:
Э то
– один из вариантов реакции переамидирования.
то
– один из вариантов реакции переамидирования.
В. Ацидолиз при получении полиэфиров и полиамидов:
Э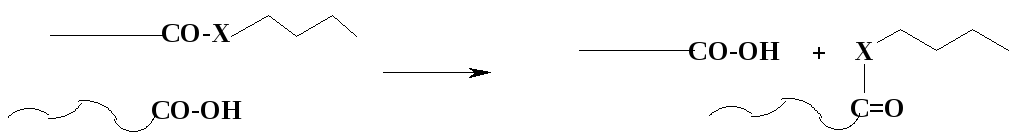 ти
реакции – другие варианты переэтерификации
(при синтезе полиэфиров, Х=О) или
переамидирования (при синтезе полиамидов,X
= NH).
ти
реакции – другие варианты переэтерификации
(при синтезе полиэфиров, Х=О) или
переамидирования (при синтезе полиамидов,X
= NH).
При высокотемпературном синтезе такие реакции протекают весьма интенсивно и по закону случая, т.е. каждая эфирная или амидная связь имеет примерно одинаковую вероятность подвергнуться деструкции. В результате образуется полимер с максимально статистическими характеристиками, в частности распределением по молекулярным массам. Если такой полимер фракционировать на ряд фракций с более узким молекулярно-массовым распределением, а затем нагреть каждую фракцию до достаточно высокой температуры, при которой начинают протекать обменные реакции, то получатся полимеры с тем же молекулярно-массовым распределением, что и исходный нефракционированный полимер. Если концевую группу такого полимера «пометить», а затем нагреть этот полимер, метка равномерно распределится по макромолекуле.
8. Стереорегулярность полимеров, получаемых путем поликонденсации, может быть достигнута использованием оптически чистых энантиомерных форм хиральных мономеров. Именно так образуются стереорегулярные (изотактические) природные полимеры: полисахариды и нуклеиновые кислоты на основе D-форм моносахаридов, полипептиды на основе L-форм аминокислот. Разумеется, при синтезе in vitro необходимо не допустить изменения конфигураций мономерных звеньев в ходе поликонденсации.
9. Конкуренция реакций циклизации и поликонденсации.
Взаимодействие функциональных групп бифункциональных мономеров может приводить не только к поликонденсации, но и к образованию низкомолекулярных циклических соединений. Если циклизация приводит к образованию 5- или 6-членных циклов, то она может составить серьезную конкуренцию поликонденсации, а в ряде случаев блокировать ее. Это связано с хорошо известной в органической химии легкостью образования 5- и 6-членных циклов; эти циклы термодинамически стабильны и образуются с достаточно большой скоростью, т.к. концевые группы цепей, из которых они образуются, расположены достаточно близко.
 Циклизация
может быть би- или мономолекулярной. К
бимолекулярной циклизации, в частности,
относятся образование лактидов (74, Х=О)
из α-аминокислот и дикетопиперазинов
(74, Х =NH)
из α-аминокислот, к мономолекулярной
– образование лактонов (75, Х=О) из γ-,δ-
и т.д. гидроксикислот и лактамов (75, Х=NH)
из γ-, δ- и т.д. аминокислот:
Циклизация
может быть би- или мономолекулярной. К
бимолекулярной циклизации, в частности,
относятся образование лактидов (74, Х=О)
из α-аминокислот и дикетопиперазинов
(74, Х =NH)
из α-аминокислот, к мономолекулярной
– образование лактонов (75, Х=О) из γ-,δ-
и т.д. гидроксикислот и лактамов (75, Х=NH)
из γ-, δ- и т.д. аминокислот:
Наиболее серьезно с поликонденсацией конкурирует мономолекулярная (внутримолекулярная) циклизация. Если в результате такой циклизации может образоваться 5-членный цикл (например, 75 при n=3), то во многих случаях поликонденсация не идет; если образуется 6-членный цикл, поликонденсация затруднена, а иногда и не протекает. В ряде случаев с поликонденсацией конкурирует и образование 7-членных циклов (например, 75, n=5).
Если предполагается конкуренция между реакциями поликонденсации и циклизации, то надо иметь в виду, что:
Повышение температуры благоприятствует циклизации.
2) Если с поликонденсацией конкурирует мономолекулярная циклизация, то ее роль возрастает при разбавлении системы, т.к. при этом уменьшается вероятность межмолекулярных столкновений, но не меняется вероятность внутримолекулярных столкновений функциональных групп (известный «принцип разбавления», используемый как раз для синтеза циклических, в том числе макроциклических структур).
В отличие от чисто органических мономеров, кремнийорганические мономеры весьма склонны к образованию 8-членных циклов, которые для силоксанов особенно устойчивы:
Такие циклы можно в определенных условиях перевести в линейные полимеры.