

3 курс / Патологическая физиология / Патофизиология_крови,_Фред_Дж_Шиффман
.pdf258 |
Глава 6. Гемостаз и |
|
тромбоз I |
мальная венозная обструкция в сочетании с повышением венозного давления являются причиной гиперчувствительности, боли, отека у больных с острым тромбозом глубоких вен.
Судьбы венозных тромбов различны (рис. 6-27). Тромб может распростра-1 няться, подвергаться лизису, организовываться или эмболизировать. Распространение тромба наиболее вероятно в том случае, если сохраняются тромбогенные | стимулы. Лизис тромба зависит от плазматического фибринолиза и лейкоцитарной эластазы, которые вызывают организацию и/или эмболизацию тромба. Как I правило, клинически значимые легочные эмболии возникают, когда тромбы образуются в крупных проксимальных венах ног (подколенной, бедренной, подвздошной), и только иногда при крупных тромбах вен голеней. Однако летальная лепи- j ная эмболия развивается вследствие как проксимальных, так и дистальных венозных тромбов.
Полный спонтанный лизис тромба происходит редко. Тромб, который не эмбо- i лизирует или не подвергается растворению, будет медленно организовываться и ре-1 канализироваться, создавая аномальную поверхность внутри вены, что приводит j к рецидивирующему тромбозу глубоких вен или постфлебитическому синдрому,
К осложнениям венозного тромбоза относятся: 1) легочная эмболия (ЛЭ) с частотой 20 на 1000 больных (ежегодно от нее умирают 142 000 американцев); 2) по-стфлебитический синдром, частоту которого трудно определить точно, но приблизительно он наблюдается у 500 000 американцев. Причина его возникновения -венозная гипертензия, связанная с деструкцией венозных клапанов, венозным
Рассасывание тромба Внутренняя
полая вена
Тромбирован
Тромбированн
Эмболия |
Организация |
Организация |
|
и реканализация |
|||
|
и инкорпорация тромба |
||
|
в сосудистую стенку |
тромба |
|
|
|
Рис. 6-27. Возможные исходы венозного тромбоза. (Из: Cortan R. S., Kumar V., Robbins S. L. (cds), Robbins Pathologic Basis of Disease, 5th ed. Philadelphia: W.B. Saunders, 1996; 108.)
Нарушения тромбообразования |
259 |
|
|
|
|
рефлюксом или стойкой обструкцией оттока крови. Постфлебитический синдром проявляется отечностью лодыжки и голени, болью в области икры и лодыжки при стоянии и ходьбе. Эти симптомы ослабевают в покое и приподнятом положении ног. Из-за высокого интракапиллярного давления эритроциты просачиваются из кровеносных сосудов и железо откладывается в коже, вызывая гиперпигментацию. Область вокруг лодыжки и голени отечна, с выраженным коллатеральным кровообращением. Иногда в средней части лодыжки возникают кожные язвы.
Наследственная тромбофилия
Дефицит AT III явился первой распознанной в 1965 г. причиной наследственной тромбофилии (называемой также состоянием повышенной свертываемости крови). В начале 1980-х гг. были обнаружены дефициты протеина С (ПС) и протеина S (IIS). На долю этих 3-х наследственных факторов риска приходится менее 15 % необъяснимых спонтанных и рецидивирующих тромбозов глубоких вен у молодых людей (табл. 6-16). В 1993 г. резистентность к активированному ПС (в результате точечной мутации в гене ФУ) была выявлена как наиболее частая причина на- следственнойтромбофилии(20-60 %). В1994 г. обнаружено, чтоещеоднимфактором риска развития венозной тромбофилии является наследственная гипер-гомоцистеинемия(19 %). В то время как дефицит AT III, ПС, US и резистентность к АПС — результат одного генного дефекта, гипергомоцистеинемия продуцируется множественными генными дефектами, поскольку различные гены осуществляют контроль нескольких ферментов, участвующих в метаболизме гомоцистеина. К другим редким причинам венозной тромбоэмболии относятся: дисфибрино-генемия, аномальный плазминоген, высокий уровень ингибитора активатора плазминогена-1 и богатого гистидином гликопротеина, аномальный кофактор гепарина II и аномальный тромбомодулин.
Виды наследственной тромбофилии
Дефицит антитромбина III. AT III представляет собой гликопротеин с одной цепью, синтезируемый печенью (период полусуществования — 65 ч). Ген AT III локализован в хромосоме 1. AT III является серпином, который инактивирует тромбин и другие сериновые протеазы (активированные факторы X, IX, XI, XII; рис. 6-10).
Дефицит AT III наследуется как аутосомно-доминантное нарушение. Оно наблюдается у 0,2-0,4 % населения. У больных с венозным тромбозом частота этого нарушения составляет 5 %. Большинство людей с этим дефектом гетерозиготны, и уровень AT III в плазме — 40-70 % от нормы. Гомозиготные больные встречаются очень редко. Тромботические осложнения у лиц с дефицитом AT III составляют от 15 % до 100 % в различных семьях. Известны 2 типа наследственного дефицита AT III. Тип I характеризуется уменьшенным количеством AT III и его дисфункцией (снижен синтез). Молекулярный дефект при типе I связан с делециями гена, кратковременными короткими вставками или делециями (мутации сдвига рамки) и единичными заменами нуклеотидов (нонсенс или миссенс-мутации).
При типе II нарушается функция AT III, но его количество нормальное (нарушенный синтез). Тип II подразделяется на 3 подтипа, которые характеризуются аномальным активным центром AT III (сайт ингибирования), аномальными сайтами связывания с гепарином, дефектом сайтов ингибирования гепарина и проте-

260 |
Глава 6. Гемостаз и |
тромбоз I
азы. Молекулярный дефект при типе II вызван миссенс-мутациями, приводящи] ми к замене аминокислот.
Дефицит протеина С и протеина S. ПС и US — витамин К-зависимые белки, син-1 тезируемые печенью. Активированный ПС (АПС) ингибирует активированные! факторы V и VIII. Т1/2 ПС равно 7 ч, а у ПБ — значительно больше (42 ч). Тромбин] медленно активирует ПС; однако тромбин, связанный с тромбомодулином (ЭК-; тромбиновый рецептор), ускоряет активацию ПС примерно в 20 000 раз. Ингиби-рование активированным ПС ФVa и ФVIIIa ускоряется посредством кофактораI nS. nS образуетмембраносвязанный комплекс с АПС, которыйоблегчает АПС | инактивацию активированных факторов V и VIII (рис. 6-11).
Недостаточность ПС и US наследуется аутосомно-доминантным путем. Гомо! зиготный дефицит ПС вызывает неонатальный тромбоз в форме фульминантной пурпуры. Распространенность гетерозиготного дефицита ПС составляет 0,3-1 0,5 % среди населения и 1-9 % у больных с венозным тромбозом. Таким образом,! большинство гетерозиготных пациентов бессимптомны. Однако полагают, что] у 50 % гетерозиготных больных из семей с симптоматическим тромбозом в анам-1 незе могут развиться тромботические осложнения.
Частота дефицита US аналогична частоте дефицита ПС у больных с тромбо-1 зом. Частота дефицита US у населения в целом неизвестна, но экстраполирование данных от когорты тромботических больных дало возможность подсчитать, что' она составляет 1:33 000. Дефицит как ПС, так и US имеет 2 фенотипа: тип I (сни-1 женный синтез) и тип II (нарушенный синтез). При дефиците ПС и nS типа I' снижены количество и функция антигена в одинаковой степени. При типе II | функция непропорционально снижена, а содержание антигена нормальное.
Синтез ПС контролируется одним геном, локализованным на хромосоме 2, а синтез US контролируется двумя гомологичными генами (а и Р), которые находятся на хромосоме 3.
При дефиците ПС типа I мутации в гене (миссенс-мутации) приводят к преждевременному окончанию синтеза или разрыву формирования вторичной структуры белковой молекулы с потерей ее стабильности. Делеции и вставки наблюдаются редко (< 10 %). Дефицит ПС типа II возникает вторично в связи с миссенс-мутациями, которые появляются в специфических местах, например домен протеазы и сайт расщепления тромбина, в результате чего возникает дисфункциональная ингибирующая активность ПС.
Мутаций гена US идентифицировано мало, возможно, из-за большого размера гена и присутствия псевдогена (р). Обнаружены большие делеции а-гена, но преобладающее число дефектов nS — это точечные мутации, которые приводят к преждевременному окончанию синтеза или нарушению вторичной структуры белковой молекулы.
Резистентность к активации ПС. Резистентность к активации протеина С является наиболее частой причиной наследственной повышенной способности крови к свертыванию. Она наблюдается у 20-65 % больных с необъяснимой венозной тромбоэмболией. Резистентность к активированному ПС связана с дефектом гена OV, который локализован на хромосоме 1. Поиск генетических дефектов для объяснения причин такой резистентности включает скрининг мутации гена ФУ, влияющей на сайт расщепления (Arg 506 — Gly 507) или место связывания активированного ПС с ФV (Arg 1865 — Не 1874). Точечная мутация в гене OV, приво-
Нарушения тромбообразования
дящая к замещению Arg 506 Gin, оказалась причиной резистентности к АПС-инактивации фактора Va. Она наблюдалась у 80-100 % обследованных больных с семейным венозным тромбозом.
Наследственная гипергомоцистеинемия. Гомоцистеин — аминокислота, образу-
ющаяся из метионина. Внутриклеточный метаболизм гомоцистеина (рис. 6-28) включает реметилирование в метионин и транссульфирование в цистеин. В реме-тилировании участвуют 3 фермента: метионинсинтаза (кофактором является ко->аламин), 5,10-метилентетрагидрофолатредуктаза и бетаин-метионинметил-рансфераза. При транссульфировании гомоцистеин трансформируется s цистатионин посредством цистатионинсинтазы (кофактором является пири-юксин). В плазме гомоцистеин окисляется в дисульфиды и смешанные дисульфиды и находится как в свободной форме, так и связанным с белком (общий гомоцистеин); нормальная концентрация в плазме составляет 5-16 ммоль/л.
Наследственные дефициты ферментов, обусловленные наследственной гипер-омоцистеинемией, включают: 1) гомозиготный дефицит цистатионинсинтазы, оторый является наиболее распространенной причиной тяжелой гипергомоцис-еинемии. Частота ее среди населения составляет от 1 : 200 000 до 1 : 335 000. ' лиц с этой патологией наблюдается высокое содержание гомоцистеина в плаз-е (> 100 ммоль/л), клинически проявляющееся ранней артериальной окклюзи-й, а также артериальным и венозным тромбозом; 2) гетерозиготный дефицит ци-гатионинсинтазы, который вызывает слабую (16-24 ммоль/л) или умеренную Ч25—100 ммоль/л) гипергомоцистеинемию. Частота встречаемости у населения составляет 0,3-1,4 %; 3) у небольшого числа больных с гомозиготным дефицитом имеют место наследственные дефекты в реметилировании, например дефицит ме-тилтрансферазы; 4) обычный дефект при реметилировании — появление мутант-нойформыметилентетрагидрофолатредуктазы, наблюдаемойу5 % населения. Приобретенная гипергомоцистеинемия также достаточно широко распространена и связана с недостаточным поступлением с пищей кобаламина, фолиевой кислоты или пиридоксина — кофакторов для метаболизма гомоцистеина. Лекарственные препараты, нарушающие обмен любого из этих кофакторов, также могут стать причиной повышения содержания гомоцистеина в плазме. Гипергомоцистеинемия вызывает артериальный и венозный тромбогенез, нарушая равновесие между антикоагулянтными и тромбогенными свойствами эндотелия и косвенно влияя на тромбоциты. Исследования in vivo показали, что гомоцистеин приводит к десквамации эндотелиальных клеток, пролиферации гладкомышечной ткани и утолщению интимы (патология сосудов). Исследования in vitro продемонстрировали, что гомоцистеин может вызывать ряд изменений в функции ЭК. Он снижает экспрессию ЭК тромбомодулина, нарушает ПС-активацию, ингибирует ТАП, нарушает продуцирование окиси азота и ППг, а также подавляет экспрессию гепаран-сульфатов. Все эти изменения вместе значительно снижают антикоагулянтные свойства эндотелия. Гомоцистеин также активирует ФУ и экспрессию тканевого фактора эндотелиальными клетками (два важных тромбогенных эффекта). Кроме того, гомоцистеин-тиолактон, содержание которого увеличивается в плазме больных с гипергомоцистеинемией, вызывает агрегацию тромбоцитов in vitro и выделение ТхА2. Этот тромбоцитарный эффект вместе с повреждением эндотелия, вызываемым гомоцистеином, объясняет механизм артериального тромбогенеза, характерного для
гипергомоцистеинемии.
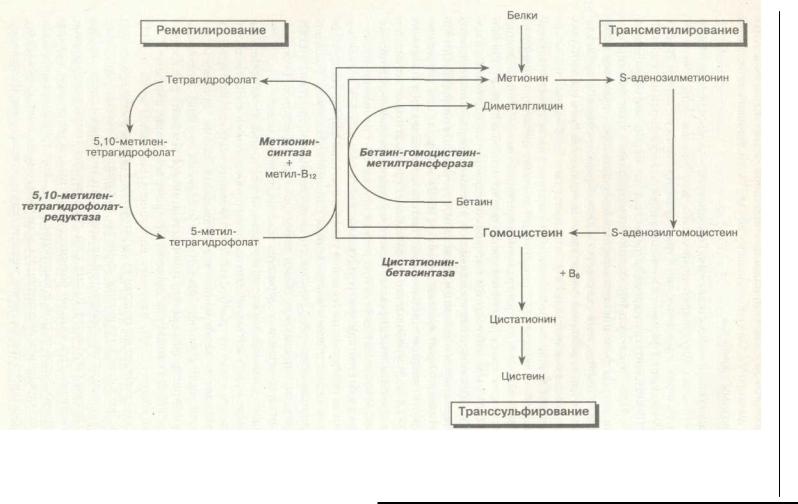
Рис. 6-28. Внутриклеточный метаболизм гомоцистеина. (Из: De Stephano V., Finazzi G., Mannucci P. M. Inherited Thrombophilia: pathogenesis, clinical syndromes and management. Blood, 1996; 87: 3531-3544.)

Нарушения тромбообразования |
263 |
Слабая и умеренная гипергомоцистеинемия является независимым фактором шска развития инсульта, инфаркта миокарда, патологии периферических сосудов и стеноза экстракраниальных сосудов. У молодых людей с венозной тромбоэмболией частота гипергомоцистеинемии достигает 19 % и в настоящее время считается фактором риска развития венозного и артериального тромбозов. Клинические проявления венозного тромбоза не отличаются от проявлений других тромбофильных состояний. Тромбоз глубоких вен и легочная эмболия наблюдаются в 64 %; поверхностные флебиты — в 24 %; тромбоз церебральных или брыжеечных вен — в 12 % случаев.
Наследственные дефекты фибринолиза. Несколько фибринолитических дефек-
тов задействованы в патогенезе тромбоза. К ним относятся: гипоплазминогене-мия, дисплазминогенемия (аномальный плазминоген), снижение выделения тканевого активатора плазминогена (ТАП) и повышенное содержание ингибитора активатора плазминогена (ИАП). Все эти изменения приводят к аномальному или сниженному образованию плазмина и нарушению (неадекватной) фибрино-литической реакции на формирование фибрина (рис. 6-13).
Наследственное высокое содержание богатого гистидином гликопротеина, который связывается с плазминогеном и препятствует образованию плазмина, обнаружено в семьях с симптоматикой тромбоза. Повышенное тромбообразование у больных с дисфибриногенемией возникает редко, как правило, вторично в связи с аномалиями фибриногена, приводящими к резистентности, лизису плазмином, повышенному связыванию с тромбином или патологической полимеризации. Генетические дефекты при первичных состояниях повышенной способности к свертыванию недостаточно полно охарактеризованы дефектами ингибиторов, описанными выше.
Клинические проявления наследственной тромбофилии
Клинические проявления у больных с дефицитом AT III, ПС, US и резистентнос-тыо к АПС сходны. Венозная тромбоэмболия наблюдается более чем у 90 % пациентов. Тромботические осложнения возникают в любом возрасте. Однако начальный тромботический эпизод обычно проявляется в раннем возрасте. Наиболее характерное место тромбоза — нижние конечности с развитием в последующем эмболии легких. Однако иногда наблюдается тромбоз необычной локализации, например в брыжеечных или церебральных венах, а также не исключен поверхностный флебит (приблизительно 5 % случаев). По неизвестным пока причинам поверхностный тромбофлебит чаще возникает у больных с дефицитами ПС и ПБ, а также резистентностью к АПС, а не при дефиците AT III.
Венознаятромбоэмболиянаблюдается у60-80 % индивидуумовсдефицитомAT III, ПСилиnS, обычноввозрастедо40 лет. УбольныхсдефицитомAT III тромбозбывает чаще, чем при дефиците ПС и US, а при резистентности к АПС склонность к тромбозу меньше, чем при дефиците AT III, ПС или US. Хотя распространенность резистентности к АПС высокая, в большинстве случаев до конца жизни течение бессимптомное. Однако резистентность к АПС в сочетании с любым другим дефектом (AT III; ПС или ПБ) повышает вероятность возникновения тромбоза.
Известно, что приобретенные факторы риска развития тромбоза (хирургические вмешательства, беременность, послеродовый период, иммобилизация) повышают частоту возникновения тромбоза у лиц с первичной гиперкоагулируемос-

264 |
Глава 6. Гемостаз и |
тромбоз |
|
тью. Так, у женщин, гетерозиготных по дефициту AT III, во время беременности или в послеродовом периоде частота тромбоза составляет 31-44 %; у больных с дефицитом ПС или ITS — 10-19 %, а с резистентностью к АПС — 28 %. Некоторые исследования выявили повышенную частоту тромбоза у больных с дефицитомAT III, ПС или nS и тех, кто подвергался абдоминальному хирургическому вмешательству по поводу опухолей. Эти клинические наблюдения подтверждают гипотезу о том, что 2 или несколько генных взаимодействий, связанных с про-тромбоэмболическими мутациями, приводят к наследственной предрасположенности к тромбозу (гиперкоагуляционное состояние), но тромботическое событие вызывается приобретенным протромботическим стимулом.
Диагноз наследственной тромбофилии
Первый этап в диагностике наследственной тромбофилии включает детальное ознакомление с анамнезом больного с учетом всех приобретенных факторов риска развития тромбоза (системная красная волчанка, злокачественные новообразования, антифосфолипидные антитела/волчаночный антикоагулянт). Необходимо выяснить возможные послеоперационные проблемы, случаи внезапной смерти в семье. Второй этап состоит в определении склонности к тромбозу: на основе собранной информации составляется схема генеалогического древа. Отрицательные показатели в семье не исключают тромбофилии, особенно резистентности к АПС и гипергомоцистеинемии вследствие низкой частоты распространенности этой патологии. Последний этап — лабораторное исследование.
Когда необходимо проводить лабораторное тестирование на тромбофилию и кого обследовать? Лабораторную диагностику нужно проводить после того, как разрешилось острое тромботическое состояние, у стабильных больных, не получающих антикоагулянты. Тестирование на определение повышенной свертываемости крови — дорогостоящая процедура. Функциональные исследования на ингибиторы свертываемости крови, проводимые в острой тромботической стадии (даже до начала лечения) неточны из-за потребления ингибиторной активности этих белков во время тромбообразования. Пероральный прием антикоагулянтов (например, варфарина) подавляет ингибиторы витамин К-зависимых ПС и Щ и стимулирует синтез AT III. Поэтому измерение их активности во время лечения антикоагулянтами дает недостоверные результаты. Лечение гепарином также влияет на функциональную активность AT III. Следовательно, лучше всего проводить лабораторную диагностику через 1-2 нед после окончания поддерживающей терапии антикоагулянтами (продолжительность обычно 3-6 мес).
Проведение лабораторного тестирования дефицита AT III, ПС и Y1S показано в первую очередь больным в возрасте до 40 лет, у которых в семье были случаи тромбоза либо этиология венозного тромбоза необъяснима. Необходимо определить концентрацию и функцию каждого ингибитора. При дефиците US следует определить уровень свободного и связанного Y1S. Резистентность к АПС и содержание гомоцистеина нужно определять у любого больного с тромбозом независимо от его возраста, поскольку распространенность этой патологиивелика. Резистентность к АПС устанавливается по способности плазмы больного противостоять пролонгированию АЧТВ, вызванному добавлением активированного белка С. Чувствительность анализа — 85 %, а специфичность — выше 90 %. Точность повышается при добавлении к тест-системе плазмы с дефицитом ФУ. Только 5-10 %
Нарушения тромбообразования __________________________________________ 265
больных с пограничными результатами требуют ДНК-анализа гена фактора V. Это исследование можно проводить уже в ходе лечения антикоагулянтами.
Содержание гомоцистеина в плазме определяют до приема метионина (внутрьвдозе0,1 г/кгмассытела) ичерез4 чпосле. Этопозволяетдифференцировать нормальное и гетерозиготное состояния при дефиците цистатионин-|3-синтазы. Для лучшего выявления гетерозиготных состояний прибегают к молекулярному анализу генных мутаций.
Лечение наследственной тромбофилии
Первичная, кратковременная, профилактика. Больные с тромбофилией (в воз-
расте до 40 лет) до, в период и после хирургического вмешательства, во время беременности, после родов (4 нед) должны получать профилактическое лечение — подкожное введение гепарина. Больные с дефицитом AT III во время беременности из-за высокого риска возникновения тромбоза должны получать или гепарин низкого молекулярного веса подкожно, или дозированный гепарин также подкожно (соотношение АЧТВ 1,5-2,0). В ходе родов рекомендуют использовать концентраты AT III (через день), а затем гепарин подкожно (в течение 4 нед после родов). Применение концентратов AT III во время беременности не дает какого-либо, в том числе экономического, преимущества по сравнению с гепарином. Гепарин с низким молекулярным весом (фракционированный гепарин) — еще одна альтернатива, но он дороже нефракционированного.
Вторичная, или долгосрочная, профилактика. Всех больных, за исключением женщин с тромбофилией, у которых развился первый эпизод тромбоза, лечат антикоагулянтами перорально (варфарин в течение 6 мес; MHO = 2-3). При множественных дефектах и рецидивах тромбоза пациенты должны постоянно принимать антикоагулянты per os.
Лечение острого тромбоза не зависит от наличия наследственного гиперкоагуля-ционного состояния. Начинают с внутривенного введения нефракционированного гепарина для поддержания отношения АЧТВ в пределах 1,5-2,5 в течение 7 дней. Как альтернатива, через день после нефракционированного гепарина больной может перейти на гепарин низкого молекулярного веса. Введение антикоагулянтов (перорально) начинают через 24-48 ч после начала гепариновой терапии и совмещают с гепарином, пока не будет достигнут терапевтический уровень (MHO = 2-3), это обычно требует 3-5 дней.
Больных с гипергомоцистеинемией лечат по поводу тромбоза, как описано выше. Профилактика пиридоксином, фолиевой кислотой, витамином В12 снижает уровень гомоцистеина, однако неясно, является ли она эффективной мерой предотвращения дальнейшего артериального или венозного тромбоза.
Приобретенная тромбофилия Послеоперационный венозный тромбоз
Послеоперационный венозный тромбоз возникает часто у пациентов в пожилом возрасте, тучных людей; лиц с венозным тромбозом в семейном анамнезе, а также после операций на брюшной и грудной полостях, головном мозге. Основные способствующие факторы: активация свертывания крови во время хирургического
266 |
Глава 6. Гемостаз и |
|
тромбоз |
вмешательства, послеоперационная гиподинамия и застой крови. Аналогичные механизмы действуют, как полагают, при застойной сердечной недостаточности, инфаркте миокарда, варикозном расширении вен.
Злокачественные заболевания
Три вида прокоагулянтов, продуцируемых злокачественными клетками, активируют или ускоряют свертывание крови и способствуют тромбозу: тканевый фактор; ферменты, превращающие ФХ в ФХа; и микровезикулы (фосфолипиды), которые служат подложкой для связывания с активированными факторами свертывания (заменители мембраны тромбоцита). Источниками этих прокоагулянтов являются аденокарциномы молочной железы, легких, предстательной железы, поджелудочной железы, тонкой кишки, при которых высока частота возникновения венозного тромбоза. Кроме того, при некоторых опухолях наблюдается неэффективный фибринолиз.
Заболевания крови
При миелопролиферативных нарушениях (МПН) и миелодиспластических синдромах, пароксизмальной ночной гемоглобинурии (ПНГ), серповидно-клеточной и гемолитической анемии возникают тромбоэмболии. При МПН гипервязкость крови, активация тромбоцитов, повреждения эндотелия приводят к активации свертывания крови и тромбозу. Основная причина смерти у больных с истинной полицитемией и эссенциальной тромбоцитемией — артериальный или венозный тромбоз. У больных с ПНГ часто возникает тромбоз печеночных вен или нижней полой вены (синдром Бадда-Киари), а также портальной и брыжеечных вен до того, как установлен диагноз ПНГ. У тромбоцитов повышается чувствительностькактивациикомплементаииндукторамагрегации. ВслучаегемолизаАДФизгемолизированныхэритроцитовиндуцируетагрегацию тромбоцитов. Для этих больных также характерен артериальный тромбоз. Серповидные эритроциты закупоривают микрососуды, а АДФ из гемолизированных эритроцитов активирует тромбоциты. Это — два важных фактора, способствующих развитию как венозного, так и артериального тромбоза при серповидно-клеточной анемии.
Лечение эстрогенами
При лечении высокими дозами эстрогенов часто возникает тромбоэмболия. Тромботические явления вторичны по отношению к повышенному синтезу факторов свертывания крови (ФН, VII, VIII, IX, X, фибриноген), снижению фибри-нолиза, ТАП, функциональной активности AT III и повреждению эндотелия. При лечении низкими дозами эстрогенов вероятность тромбоэмболии значительно ниже. Однако пероральный прием противозачаточных препаратов (низкие дозы эстрогенов) женщинами с первичным повышением свертываемости крови противопоказан из-за возрастания риска возникновения тромбоза.
Другие приобретенные заболевания
Развитию венозной тромбоэмболии способствуют нефротический синдром, воспалительные заболевания кишечника, трансплантация костного мозга. При не-фротическом синдроме больные теряют AT III с мочой, тромбоциты гиперреак-
Нарушения тромбообразования _____________________________________ |
267 |
тивны, характерно поражение сосудистой стенки. При воспалении кишечника тромбозу способствует повреждение эндотелия, приводящее к активации свертывающей системы крови и тромбоцитов, снижение содержания ПС, AT III и IS. После аллогенной трансплантации костного мозга у 21 % больных развива-тся веноокклюзионная болезнь, 45 % погибают от прогрессирующего заболевания печени. Веноокклюзионная болезнь является результатом поражения эндотелия венул и синусов печени облучением и химиотерапевтическими средствами (глава 8).
Антифосфолипидный синдром/волчаночный антикоагулянт
Определение. Антифосфолипидные антитела (АФА) — иммуноглобулины (IgG, IgM, IgA или их сочетание), взаимодействующие с одним или несколькими отрицательно заряженными фосфолипидами. Для наличия АФА характерно увеличениеАЧТВи, в меньшей степени, ПВ. Волчаночный антикоагулянт (ВА) — ингибитор антифосфолипидов, обладающий гепариноподобной активностью. Клинически АФА и ВА проявляются склонностью к развитию венозного и артериального тромбозов, но не кровотечения!
Отношение АФА к В А. Об АФА впервые стало известно в 1906 г. при серологической диагностике сифилиса. При использовании реактивов, состоящих из смеси липидов (кардиолипин, лецитин, холестерин), например VDRL для выявления сифилиса, у больных с аутоиммунными нарушениями были получены ложнопо-ложительные результаты. В 1952 г. описаны двое пациентов с системной красной волчанкой, у которых был обнаружен циркулирующий антикоагулянт (с гепариноподобной активностью), не связанный с кровотечением. Его назвали
волчаночный антикоагулянтом (ВА) — не совсем точный термин, так как у большинства больных с ВА нет системной красной волчанки.
В 1983 г. был разработан метод радиоиммуного анализа для выявления анти-кардиолипиновых антител (АКА) и было высказано предположение, что АКА и ВА — одни и те же антитела. Последующие исследования показали, что у 60 % больных эти антитела совпадают, тогда как у других пациентов было выявлено только одно из них. Некоторые больные имеют несколько антител, которые реагируют с отрицательно заряженными фосфолипидами. Эти антитела могут быть различного изотипа с различной авидностью.
Присутствие антифосфолипидных антител нередко выявляют при разных состояниях, например вирусных и бактериальных инфекциях (в частности, СПИД), аутоиммунных болезнях соединительной ткани (системная красная волчанка, ревматоидный артрит), злокачественных новообразованиях, приеме лекарственных средств (прокаинамид, хлорпромазин, хинидин). АФА обнаруживаются ч при отсутствии основного заболевания. Клиническая связь между ВА и тромбо-8ом (венозным, артериальным, отслойкой сетчатки) впервые установлена з 1963 г. у больных системной красной волчанкой. Последующие многочисленные исследования подтвердили связь между частыми случаями артериального и венозного тромбоза и АФА/ВА.
Антифосфолипидный синдром ( АФС) определяется как наличие антифосфолипидных антител (ВА или высоких/умеренных уровней антикардиолипиновых антител класса IgG или IgM) и возникновение венозного или артериального тромбо->а, рецидивирующих выкидышей и тромбоцитопении. Если основное заболевание