

3 курс / Патологическая физиология / Основы общей патологии
.pdf! |
Кальцийвданномслучаевыступапростокакэле,актролит |
321 |
какмощныймодуляторклеточнфункций,избытоккоторогох |
|
|
токсичендляклетки.Внутриконцентрациякальциялеточная |
-7 М,ч тов10000разменьше,чемв |
|
подденауровнеживается10 |
||
межклетжидк.Прифункционированиичнойстиздоровыхклеток |
|
|
прихимвнешнилиэлчхектрическихсигналов |
|
|
сопровождаетсякратк внутрышенреминымклеточнойем |
. |
|
конценкальция,чнеобходитрациидляответанастимуло |
||
Кальцийпровнчерезикаетутрьпотенциал |
-зависимвходныее |
|
кальциевканалы.Крт,ораздражениегом |
|
|
кальциймобилизующрецепторивацииведетак ф сфолипазых |
— |
|
Сипродукциилипидныхвнутриклпосредниковточных |
||
диацинозлглицеринатолтрифосфата( |
см.вышераздел |
|
«Нарушенфункцпострецепторныхияонировапосреднияковых |
|
|
механизмов»). |
|
|
Последнийвзаимодействуетембранамикальцисом вызыдепонированногоходвает [184] тамкальциявцитоплазму. Этиявленияописываются,какфункцирецепторзависимых кальциевыхканалов.
Цитоплазматическийкальцийпереходитвактивнуюформу |
|
|
|
путемвзаимодействиясосвоимбелковымвнутриклеточным |
|
-кальмодулин |
|
рецептором — кальмодулином.Комплекскальций |
|||
активируеткальмодулинзависимыепротеинкиназы,которые,вместе |
|
|
|
спрот |
еинкиназой С,активизируемойдиацилглицерином, |
|
|
осуществляютвключениетехилииныхклеточныхферментов. |
|
|
|
Характерно,чтосамикальций |
-зависимыепосредники, |
|
|
частности,кальмодулин,усиливаютпринакоплении |
|
еханизмов. |
|
внутриклеточногокальцияработууравновешивающихм |
|
||
Мощныемеханизмыинактивациицитоплазматическогокальция |
-кальциобменныйвый |
||
(АТФ -зависимыйкальциевыйнасос,натрий |
|||
механизм)быстростабилизируютприотнаветенешние |
|
|
|
раздраженияегоуровень,выбрэтоткаизсываяклеткиион, |
|
|
|
связыисекваяе |
стрируяеговкальцисомахмитохо.Одиндриях |
|
|
изклассичево,проостаещекиховраннийлпенныхриод |
— этов огпрмеждуаниос |
|
|
развитученияоповрежденииклетки |
|
||
! |
322 |
реактивным раздражением и повреждением. Где для клетки, подвергнутой внешнему воздействию, кончается одно и начинается другое? Пытаясь найти ответ, и чувствуя общее между этими процессами, И. М. Сеченов писал о «повреждающем раздражении», а Д. Н. Насонов и В. Я. Александров даже предположили, что процессы физиологического возбуждения представляют собой прообраз изменений, вызванных повреждением ткани. Согласно современным патохимическим данным, важное отличие между ответом клетки на раздражение и повреждением заключено в том, что при реактивном раздражении стабилизация уровня кальция возможна, а при повреждении емкость стабилизирующих механизмов недостаточна и концентрация внутриклеточного кальция продолжает расти и сохраняется повышенной долго.
Увеличение внутриклеточной концентрации кальция вначале обусловлено нехваткой энергии для работы кальций-магниевого насоса. Затем, при углублении гипоксии, кальций попадает в клетку не только через входные кальциевые каналы наружной мембраны, но и — особенно массивным потоком — из его внутриклеточных резервуаров — митохондрий и цистерн гладкого ЭПР, а также через поврежденные клеточные мембраны. Это приводит к критическому нарастанию его концентрации.
Показательно, что яд тапсигарджин, высвобождающий кальций из ЭПР в цитоплазму, индуцирует гибель клеток (путем апоптоза), а протоонкоген bcl-2, защищающий клетку от гибели, кодирует онкобелок, присущий цитозольной стороне мембраны ЭПР, комплексу ядерных пор и митохондриям, который удерживает кальций внутри ЭПР и перинуклеарного пространства. Блокаторы входных кальциевых каналов, а также перфторуглеродные эмульсии тормозят накопление кальция в цитоплазме поврежденных клеток и таким способом продлевают выживание клеток в условиях гипоксии.
Избыток кальция активирует ядерные эндонуклеазы, фрагментирующие ДНК в межнуклеосомных участках. Это является важным элементом апоптоза, придающим процессу запрограммированной гибели клеток необратимость. Ионы цинка служат антагонистом кальция и блокируют этот фермент, оказывая
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
323 |
цитопротэфф. екторный |
|
ПриизбыткекальцнарушаусиливаетсясинтАТФяез |
|
продукцияактивных |
кислородныхрадикаловмитохондриях.При |
высокомурокальцияклеткенеактивируетсянейтральные |
|
протеазы — кальпаины.Активностькальпаинов,доизвестной |
|
степени,можетсдерживатьсяихблокаторами |
кальпстатинами, |
запаскоторыхнебезграничен.Длительный |
избытоккальцияв |
цитоведеткплазмерогрцитоплазматическомуссирующему |
|
протеолизу.Кальпспособныразрушатьиныцитоскелет,в |
|
частности,егобелкифодринв |
-актин,чтоведетформированию |
поверхностныхвыступов( |
«blebs»)намембранегибнущейклетки. |
Кальпаинынарушаютпередачуинформациивклетке,лизируя |
С. [185] |
рецепторыипротеинкиназу |
|
Участиекальцвактвнеиявациикаскадлеточного |
|
протеолизахорошоизвестноубедительпокнпримереазано |
ихклеток |
сторполисистемыжевойплазмыКрови.Вокруггибнущ |
|
пригипоксическомнекробиозепроисходиткальцийзависимая |
|
актсистемывацияфибрина,комплеме,фибрикининов. таолиза Неисключено,чтовнутриклекаскадныепроточныееолитические
системы,таккакстемаICEигранзим |
В,известныесвоим |
участивмемханизапоптозанекроза,могутктивироватьсях |
|
избыткомкальция. |
|
Кальцийзферменттрансглутаминазависимыйможетпри избыткекальцияосуществисшивбелцитозоляк,човь способствуетформированиюапоптотическихтел цханизмам коагуляционнекрозаого,также,вероятно,формированию внутрги.алинакеточного
Активакальмембранныхцифоясфпособствуетлипаз дезинтеграцииклеточныхмемблипидныхвы ботке
медиатороввоспаления |
— производныхарахк .доновойслоты |
Этотмеханизмвнос |
итвкладразвитиеперифоквоспалеьнияого |
вочагенекробиоза. |
|
Принеобратимомповрежденииклеткимитохондрии захватываютзначительныеколичествакальц,этопр водитя инактивацииихферментов,денатурациибелков,стойкойутрате
! |
324 |
способности к продукции АТФ даже при восстановлении притока кислорода, образованию в этих органоидах аморфных электронноплотных хлопьев. Наряду с набуханием митохондрий, обусловленным проникновением в них калия, фосфата и воды и рассмотренным выше, эти процессы делают гипоксию тканевой. Такая стадия некробиоза уже не может быть обращена путем простого восстановления притока кислорода или при реперфузии.
Глубоко поврежденные митохондрии перестают быть акцепторами кислорода и субстратов. Из-за неспособности митохондрий окислять жирные кислоты их ацилы остаются в цитоплазме, где и формируют эндогенные мыла с натрием и кальцием. Омыление приводит к возрастанию детергентной активности цитозоля, который в буквальном смысле, растворяет липидные мембраны. Эндогенный детергентный эффект замыкает цепь фатальных событий, ведущих к «точке необратимости» некробиоза. Мыла разрушают мембраны органоидов и на клетку обрушивается удар гидролаз, активных радикалов и других метаболитов, изолированных до этого момента в различных отсеках клетки.
Сэтого момента, по-видимому, можно считать клетку мертвой,
апроцесс некротического аутолиза — начатым.
Таким образом, длительное повышение цитоплазматической концентрации активного кальция — это центральное звено клеточной гибели. Из-за этого нарушения запускаются несколько перечисленных выше патогенных механизмов, актуальных для гипоксического и свободнорадикального некробиоза, а также апоптоза.
Митохондрии погибших клеток продолжают быть кальциевыми ловушками и нередко становятся первичными центрами формирующегося кальциноза. Дистрофическая кальцификация происходит на месте некроза при нормальном уровне кальция в плазме.
МЕХАНИЗМЫ СВОБОДНОРАДИКАЛЬНОГО НЕКРОБИОЗА
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
|
325 |
|
Частичновосстановленные |
(активные)кислородсодержащие |
||
радикалыАКР) ( |
|
— этовысокотоксичныехимиче |
ски |
реакцимоснечетнымлноспособныекулыколичеством |
|
|
|
электр,способныеноввреждатьклеточныем ,мбраныхроматин |
|
|
|
ибелки.Отнимэлектроныу азличяорганическихмолекулых, |
|
|
|
нап, римерахк ,доновойсл кислородныебодныетырадикалы |
|
|
|
могутпреврати |
тьихвперекисныесоединениянеспаренными |
|
|
электронамизапуститьсвоегородацепныереакциивнутри |
|
|
|
клетки. |
|
|
|
АКР — физиолметаиогическиеболитыразуютсявклетке |
|
||
принормальномобменевеществ.ОнивырабатываютсяЭПРходе |
450,при |
||
работымикросомальнойокислит |
ельнойсистемыцитохромаP |
||
функционировании [186] митохосучастиемтакназываемогодрий |
|
||
убисем,влизосомахпероксихиндействиемнаомах |
-зависимыхоксидаз.Многиеважные |
|
|
мембранныхНАДФН |
|
ового |
|
процессы,напримергенерацияконечныхпродуктовпурин |
|
||
обменаираспаддофамина,обязательносопровождаются |
|
||
выработкойАКР.Врядеслучарецепторнов |
-опосредованная |
||
стимуляцияклетоксопровождаусиленипродукцииАКР. етсям |
— окисьазота |
— сама |
|
Многофунпаракринныйсигналц о альный |
|||
являетсясвободнымрад |
|
икалусиливаетомбразованиедругих |
|
АКРсм.(выше)Наконец. ,умирклеткиутоокисляютсяющие |
|
||
могутвоздейстАКРнажисоседейв.оватьых |
|
|
|
Адренэргфизиологическиаястимуляцприводит |
|
||
усилен,ахолинэргическаяю |
— кослаблениюпродукции |
-потенциал |
|
эндогенных АКР,противоположноизмедоксняет |
|||
клеткисоздаетусловиядляпермиссивногоэффекта, гдадин |
|
|
|
тотжесигналвызываетразличныйответклеток,зависимостиот |
|
||
ихредокс -состояния.Такнапример,факторнекрозаопухолей |
|
||
ФНОαвызываетлибоги |
|
бекл,льетокипролиферациюбох,так |
|
какзависимыйотнегофактортранскрипцииNF |
-квсрабатывает |
||
толькоприопресдокислительноговигееленномпотенциала |
|
|
|
клеток мишеней. |
|
|
|
АКРспособнывзаимодействоватьсульфгидрил ными группамивсоставебелков,наприме р,состаткамицистеинавих активныхцентрах,изменяябиологическиефункцииферментов
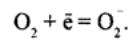
! |
326 |
рецепторов. Благодаря этому АКР могут изменять активность факторов транскрипции и тирозиновых протеинкиназ, влиять на скорость связывания цитоплазматического кальция с кальмодулином и на активность кальциевой АТФазы, на чем и основана их способность модулировать клеточные ответы на различные сигналы. Редокс-состояние клетки, по А. Я. Кульбергу, играет важную роль в управлении фазами клеточного цикла. Редокс-статус клеток находится, как и все механизмы реактивности, под генетическим контролем. Ген bcl-2, ранее упоминавшийся, как блокатор повышения цитоплазматичекой концентрации кальция, кодирует белок, присутствующий в местах основной продукции АКР и понижающий их выработку. Белок АРО-1, известный как индуктор клеточной гибели, наоборот, обладает прооксидантным действием.
Осуществляя защитные реакции, клетки, особенно, специализированные мезенхимальные элементы — макрофаги и гранулоциты — могут многократно усиливать продукцию АКР. При фагоцитозе происходит так называемый «метаболический взрыв» в фагоцитах, то есть многократное усиление потребления энергии фагоцитирующей клеткой (Балдридж, Герард; 1933). Значительная часть этой энергии расходуется НАДФН-зависимыми оксидазами на образование супероксидного радикала:
АКР осуществляют бактерицидный (цитоцидный) эффект в фаголизосомах. Так как, в отличие от лизосомальных гидролаз, АКР способны разрушать неповрежденные клеточные стенки бактерий и интактные мембраны клеток, кислородзависимый механизм завершающей стадии фагоцитоза, по современным представлениям (Бабёр, 1982), намного более важен, чем гидролитический.
Миелопероксидаза фагоцитов (в частности, нейтрофилов и моноцитов крови) превращает перекись водорода и ионы хлора в высокобактерицидный гипохлорит-анион:
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/

! |
327 |
АКРтакжесекретируютсявовнепроцессеэкзоцитоза,в |
|
|
|
|
|
расчетенаихспособностьразрушатьпричиагепервичнойный |
|
|
|
|
|
альтерации,путемпереокисмембранлениягососед их |
|
|
дениеспособствуют |
|
|
клеток,осущевторичноествляютамоповреж |
|
|
|
||
вырабэйкмедиаторовткезанвоспаленияидных.Врачи,давних |
|
|
|
|
|
временприменяющиедлядезинфекциийод,перекисьводорода, |
|
— фактически,воспроизводят |
|
||
щелокихлорнуюизвесть |
|
|
|||
эволюционносложившийсяестественныйбактерицидныймеханизм |
|
2О2,ОН —,гипохлорит |
|||
усилгентакихерациинойсоединений,какН |
|
|
|||
иоксийодиды,доступныйлюбомуфагоциту! |
|
|
[187] |
ЧерезАКР |
|
опосредуютсвоедействиефакторнекрозаопухолейидругие |
|
|
|
|
|
агенты,осуществляющиецитотоксическиеэффекты. |
|
|
|
|
|
По,индукторыстинецепныха |
|
кцийАКР |
— этоатомное |
||
оружиеклетоквборьбесносителямичужг нетическойродной |
|
|
|
|
|
информации,проникворг.Нодлющиизмтейльное |
|
|
|
|
|
значительноеувеличениепродукцииАКРведетсамоповреждению |
|
|
|
|
|
клеток. |
|
|
|
|
|
ВажнейшийизАКР |
—супероксидныйанионО |
2—.Онобразу |
ется |
||
принеполномодноэлевосстановлениикислородароннходе м |
|
|
|
|
2 под |
митохондриальнаутоокисления,такжевмикризО гомах |
|
|
|
|
|
влияниемксант,ц нтР450оидругихомаоксидаззы, |
|
|
|
|
|
показановыше. |
|
|
|
|
|
ФермсупентроксиддисмутазапревращаетО |
|
2 |
впере |
кись |
|
водор,исппротоныдальзуяНАДФН: |
|
|
|
|
|
Тотжепроцесспротекаспонтаннои , ме.дленнее |
|
Перекисьводородазникаетнетолькоизсупероксида,ноиз |
|
водыикислородаподдействиемкаталазыпероксисомах. |
|
Вприсутствиимеилдвухважелентногоза |
,образуемого |
придействиисупероксиданаFe |
+3,перекиводородараспадаетсяь |
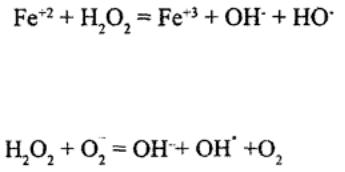
! |
328 |
образованием гидроксильного радикала (реакция Фентона):
Гидроксилы формируются и при действии супероксида на перекись водорода (реакция Хабера-Вейсса):
Радиолиз и фотолиз воды служат важным дополнительным источником гидроксилов при облучении клеток. При определенных условиях образуются и другие АКР, в частности, [188] синглетный кислород и оксигалиды (гипохлорит и оксийодид). Они служат главными микробицидными агентами фагоцитов.
Оксид азота, сам относящийся к АКР, реагируя с О2 дает супероксидный анион и перекись водорода. Окислительный стресс, вызванный избытком N0, участвует в гибели нейронов при инсульте.
Основными направлениями повреждающего действия АКР являются:
•Перекисное окисление липидов плазматической и внутриклеточных мембран, приводящее к освобождению медиаторов воспаления и токсинов (например, малональдегида, эпоксидов, эндопероксидов)
•Сшивка мембранных, внеклеточных и внутриклеточных липидов и белков через сульфгидрильные группы с инактивацией ферментов и рецепторов и образованием сульфид-радикалов, дисульфидов и сульфоновых кислот Процесс ведет к формированию белковых агрегатов (например, при катарактах хрусталика).
•Повреждение ДНК, остановка ее репликации и мутагенез, что может вызвать тератогенный или канцерогенный, а также цитостатический эффект.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
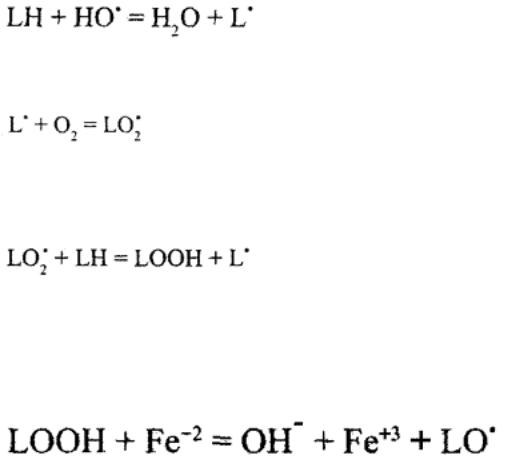
! |
329 |
Особеннопатогцепнрекисноеннокислениемембранных
липидов.
ПодвоздействиемАКРформин руетсяциирующийрадикал
липида:
Липидныйрадкислородкал |
аютлипоперекись: |
Липоператакусосмоекисьднюютлипидаекулу,формируя гидропереклипидановыйраиидсьт.д.икалный:
Двухвалентноежелезоможпревращатьгидроперекиси липидвалкосвксильободрадикалывмолярномные соотношениира1:2, зветвляяцепнуюреакциюнадва радикадообразующихпути:
Принятосчи, АКРать |
— универучастникилюбыхальные |
|
видовклеточнойгибели,покрайнмер,наееконечныхйэтапах, |
|
|
когдапроидесвнутриклеточныхкцходитмембраня |
партментовклетки. |
|
освобождениеАКРизком |
|
|
Однако,ихотн вклсительныйпратипахдзныхповреждения |
|
|
клетокнеодинаков.ПригипоксическомнекробиозеАКР,хотя |
-зависимогоповреждения |
|
образуются,например,вследствкальцийе |
- |
|
митохондрий,ноколичихнетаквелико,счтобысчво |
итатьАКР |
|
опосредоваальтерацглавмеханизмомгнуюымпоксической смертиклетки.
Правда,необход,вследзаКотраномиКумароммо(1994) отм,чтоесливтитьходеразвитиягипоксиироизошло
! |
330 |
восстановление притока кислорода к некробиотическим клеткам, поврежденные митохондрии начинают вырабатывать АКР в значительных количествах. Именно это лежит в основе так называемого реперфузионного повреждения, которое хорошо известно кардиологам. Кардиомиоциты гибнут не на высоте ишемии, а после полного или частичного восстановления коронарного кровоснабжения, будучи не в состоянии устоять перед окислительным ударом АКР. Реперфузионные нарушения диктуют необходимость комплексного лечения при остром инфаркте миокарда: врач не ограничивается мерами, направленными на лизирование тромба и вазодилатацию, а комбинирует их с введением средств, эффективных при тканевой гипоксии.
Разрушающее действие АКР является ранним и мощным и выходит на первый план в механизмах некробиоза в ряде специальных случаев, когда гибель клеток имеет определенную этиологию.
Прежде всего, это ситуации, когда резко ускоряется собственная продукция АКР — воспаление, инфекционное повреждение клеток, иммунопатологический цитолиз, разрушение опухолевых, микробных и зараженных вирусами клеток иммунной системой, трав матическии шок и синдром длительного раздавливания, а также другие состояния с активным распадом пуринов.[189]
Затем, это ситуации, когда повреждающий агент сам превращает воду и органические молекулы в свободные радикалы (радиационное поражение клеток, действие иприта и люизита, отравление кислородом и озоном, включая гипербарическую оксигенацию, гемохроматоз и отравление двухвалентным железом).
Наконец, это химическое повреждение клеток, при котором свободные радикалы формируются из молекул токсина или лекарства при их метаболизме (например, отравление четыреххлористым углеродом).
АКР особенно токсичны для клеток, богатых ненасыщенными липидами.
Генерация АКР всегда участвует в запрограммированной гибели
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/