

3 курс / Патологическая физиология / Основы общей патологии
.pdf! |
241 |
(преимущественно,гуанил ,гипок),а ственантино ферментныйбелоквразличныхслучаяпро вляет разныефизико - химичсвойтермотермостабильность( скиетвал ).
Вфункциотн,всоставенальншб ниилковм |
-ферментов |
выделяюткаталитическийуч, ллстоучастокстерический |
|
якорныйучасток,имеющиеразныфункцкодируемыеразными генами.Ураспознающихбелковнапример( ,иммуноглобулинов)
имеетсявар константнаяабельнаячасть.Патогенез |
|
наследственныхболеразкакличаетсянейприпоражениигенов, |
|
кодирующихфункциональноразличныебелки,такпридефектах |
альноразныеучастки. |
генов,шифрихункцющионх |
4Геныв. генотвзаипах,модействуюти,силуэтого, существуявлениеплейотропии — множественногодействиягена. Длянаследспатэотлогиизначаетве,чтоельзяной отожоднунарушеннуюествлятьбиохимическуюреакцодним признакомболезни.Метаболическсвязиданнойреакцимогутие
привестиктому, [139] чтоеенарушенпроявобменеится веществсразумногимиэффектамисм.также( дополнительные дананые рис. 24).
Рис. 24Нарушен. принцБидлапае |
-Тейтемаиз |
-заплейотропногодействиягенапри |
аутосомно-доминантномнаследовании. |
|
|
! |
242 |
Рассмотрим классификацию патогенетических вариантов наследственных нарушений метаболизма (по Е. Л. Розенфельду, 1980).
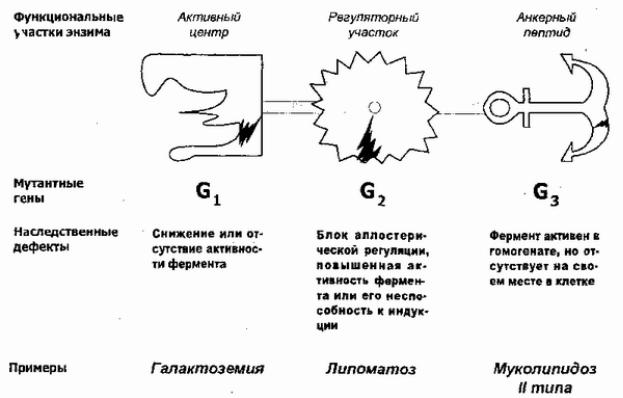
В первую очередь, коснемся наследственных болезней, поражающих белки-ферменты см. рис. 25).
Рис. 25. Виды наследственных энзимопатий
Если мутация затрагивает структуру или скорость считывания гена, кодирующего пеп-тид каталитического участка фермента, то наиболее вероятным патогенетическим последствием будет снижение или утрата активности фермента. Если активность фермента падает более, чем на 50% это проявляется клинически в виде симптомов типа классического метаболического блока, описанного выше. Примером может служить болезнь Тея-Сакса, семейная амавротическая идиотия, носительство которой весьма распространено среди евреев-ашкенази (частота мутантного аллеля до 1/30). При этом заболевании дефект каталитического участка поражает фермент гексозаминидазу А, что наблюдается во всех тканях и ведет к накоплению в ряде клеток ганглиозида Gm2.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
243 |
Еслимутациявовлекаетлостеричучастокфермента, ский предназначенныйдляеговзаимодейсрегуляторамивия ферментативнойактивност инапри( ,гор),томбазерональнаями активнфермонстражетн.Одьаэнатьделаетсяакозим нечувствительнымкаллостерическимрегуляторам.Приэтоон можувеличиватьне своюактивнсооответна тветствующиеь
стимулы.Тоедефицитгодабудет неощутимприобычном состояобменавеществ, проявитсяииприметаболической нагрузке,когдапосттрарегуляторынесмогутляционныеусилить егоактивность.
Вследстврегуляторногомутацийучасткавозможнаиутрат |
Шизвестныроколипомы |
|
способностикингибифермента. ованию |
|
|
— клональныедобр качественныепухолиизадипоцитов.Иногда |
кг)иихклеткисодержат |
|
онидостиггромадныхютзмеровдо(14 |
|
|
весьмамногожилипоматозе.Пр клеткинесутсоматическую |
|
|
мутациювгенерегуляторногоучасткафермента |
|
|
фФосфофруктокиназаазы. вмутанкле ныхках |
|
|
высо,чт,коазалбыактивна,проськлтиворессическойит |
|
|
концепцииметаблока.олическогоОднако,еслинормальный |
|
|
ферментингибируетсязбыткжирныхкислот,чтообеспечиваетм |
|
|
ограничениепереходау |
глеводовжирыпопринцотр»ицательнопу |
|
обратнойсвязи,тодефосфофрукектнаялипоматозныхкиназа |
||
клетоклишенаэтойспособн.Поэтомумутанклестиныеки |
|
|
синтезируютжирваномальнобольшихколичествах. |
|
|
Ослучайобыйпредставляетнарушениеструк уре |
гена, |
|
кодирякорныйучастокющегомолефермента.Приулыэтом |
|
|
фермпересопозннттраетнспорватьсясистклеилимойтнойки |
|
|
невзаимодействуетраспознающимбелком,кот лженрый |
|
|
заякегвнужномритьотсекеклетки.авторы( призвиненияносят |
— простоне |
|
завнез апнонахлынувшуюволнуморскихтерминов |
||
хотелосьупотребтяжеанклять«овеснп »рныйптиде |
|
|
«компартмент»). |
|
|
При муколипидозетипа2 |
— тяжеломнаследственном |
|
заболевании,проявляющемсяотставанумственномразвитии |
|
— значительносниженализосомальная |
костнымидеформациями |
|
|
активностьрядаферментов |
|
-гидролаз,втожевремяихколичество |
! |
244 |
сыворотке крови увеличено во много раз. Ферменты сохраняют свою активность и, следовательно, их активные центры не затронуты мутацией. Однако, повреждены их якорные участки, не содержащие нормального количества углеводных остатков. Углеводные остатки образуют на белковых молекулах, транспортируемых в клетке и между клетками специальный олигосахаридный адресный код, определяющий направление транспорта того или иного белка и скорость его захвата различными клетками и/или органоидами. При нарушении состава этих якорных участков лизосомы перестают узнавать и удерживать гидролазы и ферменты теряются. Дальнейшие метаболические расстройства зависят как от действия ферментов в несвойственном им месте, [140] так и от того, что субстраты этих энзимов, которые продолжают транспортироваться в лизосомы, не встречаются там с ферментами, не метаболизируются и формируют лизосомальные включения (откуда второе название этой болезни — Inclusion cell disease).
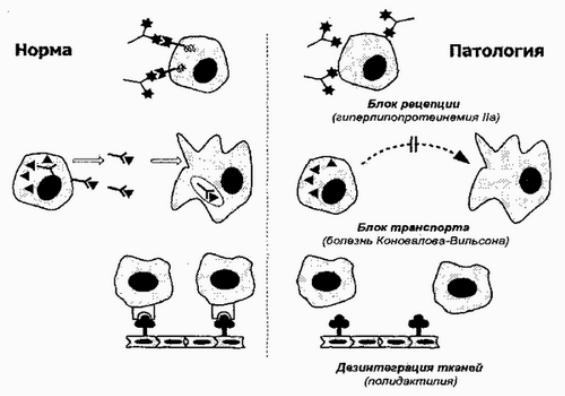
Не менее разнообразны и патогенные последствия мутаций генов распознающих белков (рис. 26).
Рис. 26. Виды наследственных рецепторопатий
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
245 |
Классприможетческимром служить-описаннаявыше разделеНа« реушенияцепциисигналов»семейнаянаследственная гиперхолесклеткииПатипа,прикотостаройновятсяемия «слепыиглухи»кпроникающемув збыткуххолестерина.
Переноссубстрачерезклемембранытоичвнутриые |
|
клетки |
— мееотсекамижду |
— нередкотребуетучасспецифическихия |
|
транспортеров — пермеаз,посутипредсобойтавляющих |
|
|
разновидностьраспознающихбелков. |
|
|
Дефектыпермеазмогутприводикнарушениюметаболизмаь |
ениясферментами. |
|
вследствпотерсубстратовилиразобщхе |
|
|
Большаяиклизнаигруппачзаболеванийескиимаяэтоготипа |
|
|
связананарушпочечногоикиниемтранспорташечного |
|
|
ами.Внокиефролоонислужосновойотрагиизвитияак |
|
— проявляются |
называемых тубулопатий.авгастроэнтерологии |
||
избиратемальабсорбциейной |
. |
|
Опистр10анаминоацидопатийспортн.изних5вызваных аномалиямигруппоспецифическнхтранспоррецептныхоров вовлекаюткаждаянесколькоблизкихпостроениюаминокислот.
Цистинурия,болезньХартнупадибазикаминоацидурия еют клизнаипроявленияческиимые,адикарбоксиламиноацидурия иминоглицинурия — бессимптомны.Другиетранспортных5 аминоацидопат:гиперцистинурия,гистид,лизинурия. й мальабсорбциятриптофаметионина
Всеэтисостояния наследственныеболезни.Наиболечастаклизнаическиима цистинурия (1/15000)Цистинмалорастворимприданном. заболеванииформируеткамнипочек,мочевогопузыряуретры.В
дополнениек [141] этому,больныхснижено кишечноевсасывание иувеличенапочечнаяэкскрецияорн,л аргининатз ,не толькоцистина.
При болезниХартнупа |
(1/24000)нарушеновсасывание |
увеличенаэкскрециянейтральныхаминокислот,,вследствие |
нтеза |
нехваткитриптофанаразвиваетсянарушениеси |
! |
246 |
никотинамиддинуклеотидфосфата — энергетического носителя, являющегося производным витамина РР, и формируется пеллагра, как и при авитаминозе по данному витамину.
При дибазикаминоацидурии (1/60000, относительно часто бывает у финнов и франкоканадцев) не реабсорбируются аргинин, орнитин и лизин. Камнеобразование для данного заболевания нехарактерно, но у больных развивается нарушение цикла мочевины, гипераммониемия и задержка роста.
Триптофанмальабсорбция характеризуется повышенным выделением индолов с мочой и калом, так как невсосавшийся триптофан становится добычей кишечных бактерий, что приводит к аутоинтоксикации. Наблюдается кальциноз почек.
При метионинмальабсорбции — дефицит метионина проявляется умственным отставанием, задержкой роста, отеками, приступами одышки и судорогами. Как правило, из-за нарушения образования меланина у больных светлые глаза и волосы.
Иногда дефект распознающего белка приводит к нарушению функций того или иного сывороточного переносчика. Именно к этой группе заболеваний относится болезнь Коновалова-Вильсона (1/30000) или гепатолентикулярная дегенерация. Первичный дефект при данной болезни касается 13 хромосомы и, по некоторым предположениям, состоит в мутации гена, кодирующего экскреторный переносчик меди, позволяющий печеночным клеткам выделять ее избыток в желчь.
Вследствие этого используется (или не синтезируется в достаточном количестве) церулоплазмин, переносящий медь из печеночного депо в ткани. Ведущие проявления заболевания связаны с постепенно формирующейся печеночной недостаточностью и некротическими изменениями в ЦНС, практически, по [142] типу эндогенного отравления тяжелыми металлами, так как избыток меди задерживается в поражаемых тканях.
В течение многих лет принято было выделять отдельно наследственные болезни, связанные с дефектами так называемых структурных белков. На наш взгляд, оправданно их причисление к
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
|
|
247 |
|
группедефраспктбелковзнающих |
|
— вмеспатологией |
||
рецеп,пермеазитранспортныхоровбелков. |
|
|
||
Деловтом,чтостроифункция,тестьельнаяоучастие |
|
белкав |
||
самосборкемежклеточноговещ,либокакихства |
|
-клеточных |
||
структур,опредвсеспособнгоцелоляетсяраспознаватьстью |
|
|
||
какие — тодругиеструктурэлемеилираспознаватьсятыыеими. |
|
|||
Примераминаследственныхболезней,принадлежащихкданной |
|
синдром |
||
группе, |
могслужить |
коллагенопатии.Такназываемый |
||
Черногубова-Элерса-Данло,характеризующсистемнымийся |
|
|||
нарушениямипрочностструктурыколлагена,находитсвое |
|
|
||
выражениеповышрастяжимостисниженнойпрочнойсти |
ав,ломкостивсосудов,а |
|
||
кожи,излишнейподвижностисуст |
|
|
||
некоторбольныхосложняеразрывамивнуоргановтреннихся |
|
|
||
(артерий,матки,кишечн)Трад. считавшийсякациоединымно |
|
|
||
нозологическпоняти,развитм мдицинскойгенетикием |
|
ных |
||
данныйсиндоказалсяразноромгруппмоднойген |
|
|||
наследствзабол,связаеннныхрушениемметаболизманыхий |
|
|
||
колл.Внастоящеегенавремявыдменееляют11типовданного |
|
|
||
синдрома,вызванныхдефектамиразличныхген.Повдобная |
|
|
||
гетерогенностьобуслпр такявлениемвленаназываемых |
|
|
||
генокопий. |
|
|
|
|
Иногда,рассматриваяродосбо,медикильныховныесталкиваются |
|
|||
тем,чтоприодинаилисходнойклиническойовойкартинеболезни |
|
|
||
типеенаследованияуразныхпациентовразличается.Этосвязано |
|
|
||
тем,чтоодноитожеметаболичнарушможетбытьниеское |
|
|
||
результатомсрываразличныхзвеньевметаболическойцепи, |
|
|||
феирментыецепторы,обес оследовательныеечивающиешага |
|
|
||
выподобнойлнепрограммы,кодируютсяииразличнымигенами, |
-разному. |
|
||
расположеннымивген ме |
|
индрома |
||
Так,празличныхвариантахрассматриваемого |
|
|||
клиническаякартинаможетвызыватьсядефектомбиосинтезаили |
-Данло,тип3), |
|
||
структурыпроколлагтипа4синдром( Эленарса |
|
- |
||
дефектомлизил |
-гидроксилазытип(деф6),мектомдь |
|||
транспортирующейАТФ |
-азытип),дефектом(9 |
|
||
коллагенраспознающегабелкафибр |
онектина(10тип),нарушением |
|
||
превроколлагенаащениятипа1в тип(при7)этом |
|
|
||
последняяаномалия,всвоюочередь,гетерогеннаиможетбыть |
-N-протеилиженазы |
|
||
обусловленадефектпрок мллаген |
|
|
||
! |
248 |
ошибками в структуре генов ее субстрата. При этом наблюдаются различные типы наследования — так. 1-4 и 8 типы синдрома аутосомно-доминантны, 5 тип рецессивен и сцеплен с Х- хромосомой, 6 и 10 — аутосомно-рецессивны, а 7 тип имеет и аутосомно-доминантный и аутосомно-рецессивный вариант.
Данный пример убеждает в относительности патогенетической классификации наследственных болезней — ведь при коллагенопатиях одни и те же клинические симптомы возникают и при дефекте распознающих белков (тип 3, 10) и при дефектах ферментов (тип 6). Практический вывод из существования генокопий — невозможность дать квалифицированную медико- генетическую консультацию на основании только клинико- лабораторной картины болезни у пробанда и безусловная необходимость генеалогического исследования.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
249 |
ГлаваRПОВРЕЖДЕНИЕ8.
ИСПОЛНИТЕЛЬНОГОАПП АРАТА КЛЕТКИ
Досихпормыобсуждинформациочногоыеаспекликлеты поврежде,связнанрушнсиыеягнал,воспрниямизацииятия передуправсигналовчи,такжеяющихдефектами адаптационпрограммклетки.Однакопроблемаыхклеточного повреждения имеетдругую,оченьважнуюсторону.Повреждение приводитктиповымизмененразличныхотсекаклеткиям. Данныйразделпосвященовременнымданнымпатохимических законповреждениямерноклеточногос«полнительноготях
аппар»Вн. мырассмотримчтале ихпоотдельности, применительнокразличорг,затемнелламопишемым интегральныемехразличныхнизмыспособовгибеликлетки.
ПАТОХИМИЧЕСКПОСЛЕДСТВИЯЕ ПОВРЕЖКЛЕТОЧНОГОЯДРАЕНИЯ
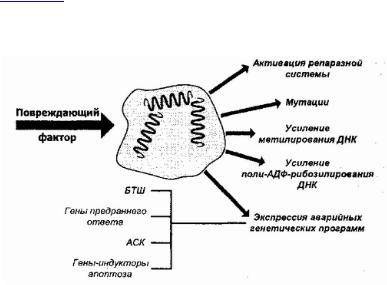
ПовреждениеядернойДНКвызываетнесколькотиповых защитныхреакций.Одн а,коаждыйизэтихмеханизмов «погрешим»,срабаты,станопричинойваяторичныхится нарушений( рис. 27).
Рис. 27Типовыепоследствия. поврежклеточного. раения БТШ — белкитеплшока, вого АСК — антигенстареющихклеток
! |
250 |
Рассмотрим это на конкретных примерах. Выше уже шла речь о системе эксцизионной репарации пиримидиновых димеров и однонитевых разрывов ДНК и о потенциальных опасностях, связанных с ее несовершенной работой.
К этому можно добавить, что эндонуклеазы, играющие важную роль в работе репаразной системы, при определенных условиях могут обеспечить начало апоптоза — запрограммированной гибели поврежденной клетки с фрагментацией ее ДНК. Более того, подобный «вызов огня на себя» для устранения повреждения вместе с клеткой, которая его несет, по-видимому, является достаточно характерным результатом повреждения ядра, так как очень многие повреждающие агенты индуцируют апоптоз клеток-мишеней, а конституциональный элемент системы препарирования мутаций — ген р53, упоминавшийся выше, как «замораживатель» митотического цикла поврежденных клеток в G1-фазе, известен также как ген-индуктор апоптоза.
Достаточно типичным биохимическим последствием повреждения хроматина служит усиление метилирования и АДФ- рибозилирования ДНК и ядерных белков.
Известно, что в хроматине активные гены менее метилированы, поэтому первая из данных биохимических реакций может отражать инактивацию во время клеточного повреждения ряда «неэкстренных» текущих клеточных программ, которые на период острой адаптации к повреждению архивируются.
Что же касается роли и последствий усиления поли-АДФ- рибозилирования, то эта сторона ядерного ответа на повреждение может весьма драматически влиять на судьбу поврежденной клетки. В присутствии поврежденной ДНК активируется фермент поли- АДФ-рибозилполимераза (ПАРП). Фермент превращает остатки НАД в поли-АДФ-рибозу и вызывает сшивку белков хроматина через поли-АДФ-рибозильные мостики. Данный [144] механизм является универсальным для всех живых клеток. Он направлен прежде всего на закрепление концов разорванной хроматиновой нити и, фактически, участвует в работе репаразной системы, склеивая разрывы, препятствуя транскрипции поврежденной ДНК и,
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/