

3 курс / Патологическая физиология / Основы общей патологии
.pdf! |
221 |
генетиковопас,чтониеловеческаяпородавследствиеэтого деградирует.Основателемантропологическойгенетики Ф. Гальтоном(1822 -1911)былпредложентерминевгеника« »для
обознанаучноения -практическойдоктрины,улучшающей человеческуюпородупутемискусственногоотбора.
Упртощенноелкованиеза оновлассичесгенети, киой |
|
|
|
лигенном |
|||
существовавшеедоформировпредстпо ниявлений |
|
|
|
||||
наследовании,создаугенетикалопе,чслабоумиетоатлениев |
|
|
|
|
|
||
другиедезадаптирующиенаследственныесиндромымоног нны |
|
Паннетт, 1917;Г. |
Годдард1917). |
|
|||
подчиняютсязаконамМенделяР.( |
|
|
Ист, |
||||
Всвязиэт,видныегенетикимтоговремениЭ. ( |
|
|
|
М. |
|||
Р. А. Фишер, X. |
Нильсон-Эле,. |
Бауэр)пришликвыводу |
|
||||
возможсильноуменьшитьчастэтибохптулезнейтем |
|
|
|
|
|
||
стерилизацииограниченразмножносителейяния |
Давенгоду1912портлагал |
|
|
||||
патологическихгенов.Ч. |
|
заоднопоколение |
|
||||
возможнымизбавитьсяотдефгеновктных |
|
|
|
|
|||
генетизоляцителейихночес.Р.кой |
|
|
|
|
Панне,вчас, тности |
|
|
считалвозможнымевгеническимиметодамиобеспечитьредукцию |
|
|
|
|
|
||
часлабоутотысоскоростьюнеменееия11%заоднопоколение. |
|
|
|
|
чную |
||
Евгеническиеидеипр этотобрелиперш науодрокую |
|
|
|
|
|||
известнопопулярностьобще .Таквенную, году1918 |
|
|
|
|
|
||
П. ПопеноэиР. |
Джонстонписали:Лишь«немногиев ашевремя |
|
|
|
|
||
согласятсятем,чтодвумслабоумнымилиэпилептикам |
|
|
|
[127] |
свою |
||
принекоадлженправо«» иувековечитьься |
|
|
|
||||
породу, этому |
|
уточкузрениянестоитдажеоспаривать»В. |
|
|
|
||
доклаАмерикансго1914да обществуселекционеровому |
|
|
|
|
|
||
Г. Лафлинвыразилкредоевгеникитак: Общество"должн |
|
|
|
|
|
||
рассматриватьзародышевуюплазмукакобщественноедостояние, |
|
|
ыйявляетсяееносителем». |
|
|||
непростособственностьиндивида,котор |
|
|
|
|
|||
ПодобнойстепениобобществлениянетребовалдажеМанифест |
МарксаиФ. |
Энгельса(1848),авторы |
|
||||
коммунистическойпартииК. |
|
||||||
которогопряотмежидобщн«евалижен»!Вотуж,стиь |
|
|
|
дахидпеярешла |
|
||
поистиевгенический, социализм.Втридцатыхго |
|
|
|
|
|||
вплоскостьпрактическойреализации.Стерилбы зация |
|
|
|
|
|
||
узаконенавнацистскойГерман(1933),вБрКолумбииганской |
|
|
— |
с 1929,адля«пользы |
|
||
(Канада, 1933),вДаниидобровольно |
|
|
и |
||||
общества» |
— с1934),вНорвегии(1934),Швец1934),Финлянди |
|
|
|
|||
(1935),Эстонии(1936),Исландии(1938)Всоргоковыхдах.
! |
222 |
активную государственную евгеническую политику проводили нацисты. Интересно, что, по данным У. Дейчманна и Б. Мюллер- Хилла (1994) среди ученых Германии наибольший процент членов национал-социалистской партии был именно у представителей медико-биологических наук.
Лишь немногие видные генетики открыто и последовательно выступали в эту эпоху против модной евгеники. Дж. Б. С. Холдейн называл ее «образчиком ярого американизма» и указывал на то, что закон Харди-Вайнберга предсказывает столь медленный темп устранения гетерозигот из популяции, что с надеждами добиться евгеническими мерами значимого эффекта для общественного здоровья в реальные сроки можно расстаться. К этому можно добавить мультифакториальность наследования большинства распространенных серьезных заболеваний, возникновение мутантных аллелей в популяции заново, и, наконец, соображение о продолжении действия отбора в популяциях человека, приведенное выше.
Аллели уникальной потенциальной ценности могут находиться в разнообразных сочетаниях с патологическими генами у гетерозигот, поэтому попытка быть умнее природы может стоить популяции потери многих ценных генов.
Когда все эти соображения были оценены медиками, евгеника вышла из моды, и возобладало убеждение, что «размножение должно оставаться частным делом в контексте уважения медицинских прав пациента» (Д. Пол, X. Спенсер, 1995). Если генетика — основа индивидуальности человека, значит только индивид может, в меру своего естественного несовершенства и неизбежной погрешимости, правильно решать вопросы судьбы той части генофонда, которой он располагает (или, лучше сказать, которая располагает им!).
Возможно, открытие в 90-х годах аллелей, связанных с высоким интеллектом и, наоборот, предрасположенностью к таким социальным недугам, как наркомания, обострит интерес к евгенике вновь. С точки зрения основной проблемы, обсуждаемой нами — проблемы несовершенства организма — евгенические устремления
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
|
|
|
223 |
|
психологичеспонятны.Этопопомощьюглавноготки |
|
|
|||
преимуществачеловека |
— интеллектаисправитьтупогрешимость, |
|
|||
котораяорганпрбиологиисущачески |
|
Homo sapiens. |
|
||
Наслдедственныефекты |
- |
результатповреждениямутагенами |
|
||
ДНКсм(. |
рис. 19). |
|
|
|
|
|
|
|
|
|
|
Наиболееважн |
ыемутагены |
— химическиенаприм(, |
р |
||
интеркалирующиеаген,ци ),ыостатикифизическиефакторы |
|
|
|||
(радиация),биологагентыв(и)руче. скиеы |
|
|
|
||
Вместетем,какприлюбомзаб, леваниидногодействия причиннфактонедостаточнодляргоазвитиянаследственной болезни.Необхоопределенныеуслимывияответствующее состоянреакторгаи.евнизмаости
Мутацияещенеозначаслзаболедствинееваниянного равнозначнаэтомупонятию.
Вышепрассмотренииобщейэтиологииболезнейуже отмечалось,чтоусловияреакт ивностьорганизмагуттменить действиепричфактораразвитиеннболезниго.
Этосправедливодляболезннасл,развитиийдственных |
[128] |
|
которыхмутациянеобх,нодостаточнадима. |
||
Во-первых,биологантическиезащитныемутационные |
|
|
механизмы,прис |
ущиереактздоровыхивностиндивидов,способны |
|
исправлятьилинейтрализмутации,либоликвидировать мутанкле. тныеки
Во-вторых,генетическийаппароблопределеннойадает надеж,всилучегонкаждоеостьюповреждениеструктурыДНК оборачиваетсяклин ическизначимымипоследствиями.
В-третьих,выявлениемногихмутацийвозможнотолькопри |
|||
перехпоруровнядегздействиягоопределенныхфакторов |
|||
внешнейсреды.Так, |
алактазия проявляетсявидерасстройства |
||
кишечникалишьутехгомозиготныхносителейдеф |
|
|
ектногогена |
ла,которыетазыпьютмолоко( |
|
рис. 20). |
|
|
|
|
|
! |
224 |
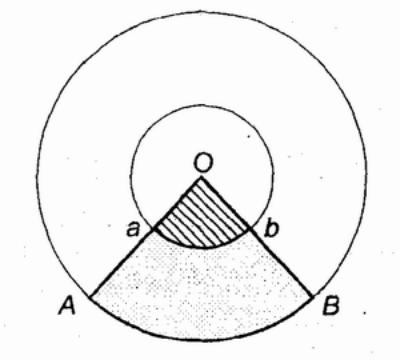
ис. 20. Взаимодействие мутаций и наследственных болезней.
нешний круг — носители дефектного гена. Внутренний круг — части носителей мутации, одверженных действию экзогенного патогенного фактора. Сектор АОВ — часть носителей мутации фенотгтической аномалией. Сектор аОв — носители клинически выраженного заболевания.
И, наконец, в-четвертых отбор генов и дрейф генов могут устранять мутантный аллель из популяции, предохраняя следующие поколения от болезни.
Определенные предпосылки стабильности и консерватизма генома связаны с самой его структурой. Двойной характер спирали ДНК расценивается молекулярной генетикой как фактор увеличивающий надежность хранения генетической информации, так как при однонитевом повреждении сохраняется возможность восстановить программную запись по ее антисмысловой реплике.
Широко известно свойство вырожденности. присущей генетическому коду. Кодоны, отличающиеся между собой в третьем знаке, шифруют одну и ту же аминокислоту. Например, после двух остатков цитозина (СС*) третий нуклеотид может быть любым — и система белкового синтеза все равно читает кодон как пролиновый и синтезирует нормальный коллаген. Не будь генетический код вырожденным, количество мутаций, дающих наследственные болезни было бы существенно большим.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
225 |
Диплоидностьсоматическихклетокорганизматоже рассматриваетсякакфакгордополнительнойнадежности. БлагодаэффектуМэЛайон,рвдиплоидныхя соматическихI клетактивна, правилох,однаизкаждойпарыгомологичных
хромосом.Какаяименно — отцовскаяилиматеринская определяетсяпослучайпринципу.Привысокойомустепени гетерозигот,присущейбольшиндиви,этотисонствуздаетов дляпуклдопоаеток лнительныйвыбордоступныхпрограмм адаптации.Еслиоднаизхромосомнесетдефектныйаллель,то,по крайнмервполовинеклетй,произошоткданнойкл,еткидших работаудеегон трманалогь. ьный
Неслучайнобольшинствонаследственныхэнзимопатий рецессивны:одинмутантныйгеясоздаетлишьдефицит50% фер,чтомобытьежнетаощутимо,таккакдлямножества фермеорганизмнтативныхакцийсостоянииподнять концентрациисубстрвраза2восстановитьтовнормальную
производительностьданногометаболи ческогопути,вцелом. Имепоэт,нноапример,гетерму носзителиготные фенилкнеимсимптомовеюттонурииболезниДж.(
М. Браун. 1994).
ВслучаеповрежденияядернойДНКвступаютдействие биохимичемеханизмыеевосстановления.Оникиерезаю поврежденныйучасток,восстанавливаядолжную последовательностьДНКматрицекомплементарной, писли последняяохранена.
Данныйпроцесссложенитребуетучасчеэнзимовырехия: эндонуклеазы,вырезающейповрежденныйучаст,экз, нуклеазы разрушающей [129] дефце,ДНКктнуюпь синтезирующейвосстановпоследовигенную, азытельность вкленаивающейместо.
ПомнениюДж.Хофмана(1990),этотзащитныймеханизм достатмощенипозволяетисправитьчнодоспонтанных95% мутаций,включаяодно нитдефектыивые,возможно,наименее тяжелыедвунитевыеповрежде.Однако,при ниятевых разрывахегодейсзатотсутствиемрудненоантисмысловой
—
Гольдштейн,
т
-полимеразы,
! |
226 |
матрицы. К тому же, не исключено, что работа данного механизма не всегда приводит к прецизионной сшивке вновь синтезированного фрагмента ДНК и остатков старой цепи «конец в конец». Если же при этом происходит какая-либо транслокация фрагмента ДНК, то подобное событие само может привести к изменению экспрессии генов и нежелательным последствиям — именно путем транслокации активируются некоторые протоонкогены, например с- myc, ответственный за развитие злокачественной лимфомы Беркитта.
Несмотря на свое несовершенство, репа-разный механизм представляет собой заслон от мутаций, значение которого трудно переоценить. Сама природа убеждает в этом с помощью трагических примеров наследственных болезней, при которых восстановление повреждений ДНК в тех или иных клетках не осуществляется или понижено.
Подобные нарушения объединяются под названием синдромов хромосомной нестабильности. К ним относятся пигментная ксеродерма, врожденная аутосомно-рецессивная апластическая анемия Фанкони, атаксия-абазия-телеангиэктазия и синдром Блума.
При всех этих заболеваниях репаразная система имеет ферментативные дефекты, в частности, при анемии Фанкони и пигментной ксеродерме — дефект гамма-эндонуклеазы.
Результатом является многократное увеличение частоты соматических мутаций. У больных выявляются клоны клеток, несущих хромосомные аберрации. Летальные мутации обуславливают клеточную гибель, которая, например, вызывает аплазию костного мозга, малокровие, лейкопению и тромбоцитопению у пациентов с анемией Фанкони.
Резко снижается толерантность к мутагенам, например, при пигментной ксеродерме — к ультрафиолетовым лучам. Значительно возрастает вероятность озлокачествления и опухолевой трансформации клеток. Так, при пигментной ксеродерме резко повышается риск рака кожи, анемия Фанкони и атаксия-телеангиэктазия считаются предлейкозными
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
227 |
заболевания,присиндроБлумачавстречаютсяиовсе наиболеераспрострформырака,вособенности,ненныеорганов желудочно-кишечноготракта.
Предп,чтдефектыорассматриваемоголагают антимутехслужатодниммацфакторовонногозпат генеза проге,сопровопреждевременнымемойии ста.рением
Еслимутация |
|
небылаисправленабиохимически,возник |
|
|
мутантныйклонклеток,дальнейшееразвитиболезниможетбыть |
|
|
||
предотвпутемостановкиразмножщедефеккл.етнияныхок |
|
|
||
Одновременносактивациейрепараз,клетках,претерпевших |
специальныегены, |
|
||
поврежденДНК,активизируютсяе |
|
|||
вызывостановклеточногоющиеци.Одинизпродуктовла таких |
|
|
||
генов — белокр53 |
.Мутации,поражающиегенэтогобелка,нередко |
|
||
обнаруживаюопухолевыхклеточныхклонах,чтося |
|
|
|
|
свидетельствуетоважнойзащитнойролизапрограммированной |
|
|||
остановкиразмножпредотвращениядефекклдляетныхок |
|
|
||
онкологическзаболеван. ихй |
|
|
|
|
Еслимутантныйклонпродолжаетпролиферировать |
— встает |
|||
задачаистребленияклетанопрограммальнымкаппарато. ным |
|
|
||
Этузадачурешаетиммуннаясистема. |
|
|
|
|
Мутанклеткип ные |
|
одвергаютсяатакеспецифически |
- |
|
сенсибилизированныхопухолевымиантигенамиCD |
-8 |
|||
положительныхлимфоцитов |
|
-киллеров,проти оопухолевых |
|
|
антител,стимулированныхантителамимакр рмальныхфагов |
|
|
||
киллеров(NK |
-клеток). |
|
|
|
Иммунологич,механизмустранениямут ский |
ацийменее |
|
||
знач,чембиохимический.Уносителейврожденных |
|
|
||
иммунодефицитовчасткл заболетальных,вызванийных |
[130] |
|
||
мутациями,возрастаетсотнираз. |
|
|
|
|
По-видимому,егор льсобенновеликаборьбе последствсоматичмутаций,хотянямиеских исключено,что спонтаннвыкидплодовпослышиегедствияминеративных мутацимеют,отчасти, йммунологмеханизм. ческий
! |
228 |
К сожалению, мутантные клетки, например, опухолевые могут ускользать от иммунологического надзора. Какие же принципиально могут быть повреждения в структуре ДНК? Виды мутаций разнообразны.
Минимальное мутагенное повреждение ДНК связано с заменой одного азотистого основания на другое. Это так называемые точковые мутации. Одно или несколько азотистых оснований могут быть помещены в нормальный ген (вставка) или утрачены им
(делеция). Выделяют инверсию участков генов, их транслокацию
на другие гены, а также дупликацию.
Смысловые последствия этих мутаций для исполнения клеточных программ белкового синтеза могут быть различными.
Миссенс-мутации происходят в генах, кодирующих полипептиды (экзонах) и заключаются в нуклеотидных заменах, приводящих к синтезу полипептидных цепей с аминокислотными заменами. Классическим стал пример серповидноклеточной анемии, первой болезни, которую Лайнус Полинг назвал «молекулярной». Миссенс-мутация с заменой кодона СТС в гене β-глобина на САС ведет к подмене глютаминовой кислоты в β-глобиновой цепочке гемоглобина S — валином.
При нонсенс-мутациях экзонов кодон аминокислоты меняется на один из стоп-кодонов — особых знаков препинания в генетическом коде, терминирующих трансляцию.
Так, замена в том же гене β-глобина кодона глютамина CAG на стоп-кодон UAG преждевременно остановит синтез бета-цепи глобина и последняя разрушится. Результатом будет синтез гемоглобина, лишенного бета-цепей, что ведет к особой наследственной гемолитической анемии (бета-ноль талассемии).
Интроны или некодирующие последовательности в геноме эукариот перемежаются с экзонами и содержат «служебную информацию», необходимую для работы ядерных ферментов и биорегуляторов с генами внутри ядра, в частности, промоторы и энхансеры. разрешающие или усиливающие транскрипцию соседних экзонов. Интроны списываются при первичной транскрипции и их
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
229 |
копиинаходявсоставеге сяерогенныхядерныхРНК,нопри редактированиипоследнихпроисхвырезаниекинтроновпд й сшивсплайсинг( )копийэкзоновединформационнуюРНК. Таккакэ частозоныкодируютпоследовательныепептидыиз составаодногобелка,точноевыринтронзаниесплайсингмогутв бытьоченьва жныдляправильнойреализациипрограммыбелкового синтеза.
Мутацииинтро,казалосьбы,нзатреовнагиваютпрямую генетичекодбелков.Однако,заскийчетнарушения вышеописаннредактипервичованияанскриптаонигогоже
могбуславливатьнаследств енныебол.Внешнеэтознивыглядит какизменескорсинтопределенныхиестиполипептидовза.
Именнотакмутацииинтроновответственнызанаибольшее
числослучаевбета |
-талассемии.Примутированиипромотора |
интранскрипцияонеданногоэкзонабудетдав |
лена. |
Примутирместаоедваниинитронасэкзономнения сплнарушйсингбетается -глобин,матричнаяРНКкоторого должнабытьсшитаизтрехфрагмеэкзокопий,новыхтове синтезирувообщбета( ется -нольталассемия)Прилокализации.
поврсежденияредине интамсоздаютсяроновыеточки прикреферментовсплайсинга,енияпараллельнопроисходят нормипатологическльныйсплайсинг.Приэтомразвиваетсяй бета-плюсталассемия.
Приделециивставкелихотябыодногонуклеотидапроисходит сдвиграмкисчитыв ания,тоестьпослекобдуоныдутющие
читнатьсячинвстнуклеотидавлеяилинуклеотиданого, |
|
|
следующегозаделетир.Приэтнарушитсяваннымтрансл, ция |
|
|
вполипептиднойцепимогутпоявитьсязаменынескольдущих |
циальноесчитываниепривелок |
|
подрядаминокислотили,е липе |
||
созданиюстоп |
-кодона,наблюдобтранскрипцииывцепиется.Итак |
|
мутациимогутнарушатьтранскрипцию,слайсиили, сляциюг |
|
|
прекращатьилипродсинтезолжитьпредебелкаилиенного |
мкамивпервичной |
|
приводитьксинтезубелковыхмолекулполо |
||
структуре. |
|
|
Средиазвивающихсярезультатемутацийнаследственных
! |
230 |
болезней традиционно выделяют три подгруппы.
Моногенные наследственные заболевания представляют у каждого пораженного индивида результат мутации одного гена. Так как каждый такой конкретный ген имеет строго определенную локализацию в той или иной хромосоме и определенные свойства, все мохромосомные заболевания подчиняются законам классической генетики, сформулированным Г. Менделем (1865) или, как говорят, «менделируют». Для моногенных наследственных заболеваний , в том случае, если мутация не возникла впервые у родителей пациента-пробанда генеалогическое исследование выявляет один из трех типов наследования: аутосомнодоминантный (когда признак фенотипически проявляется даже у гетерозигот — при наличии в неполовых хромосомах хотя бы одного мутантного гена), аутосомно-рецессивного (когда мутантный ген, локализованный в неполовых хромосомах, находит фенотипическое проявление только при условии гомозиготности — если в искомом локусе оба аллеля мутантны), или, наконец,
сцепленное с полом наследование (когда мутантный ген находится в X либо в Y-хромосоме). Примеры родословных, типичных для этих видов наследования, отражены на рис. 21.
Наследование, сцепленное с полом нельзя смешивать с явлением ограничения экспрессии определенных мутантных генов полом. Так. ген почечного несахарного диабета, обусловливающий резистентность клеток собирательных трубок нефронов к действию вазопрессина, локализован в Х-хромосоме, в связи с чем данная болезнь наследуется рецессив, [131] но, сцепленно с полом, поражая почти всегда мужчин.
Облысение, как известно, тоже мужская болезнь. Среди женщин случаи алопеции так редки, что заглавие известной пьесы Эжена Ионеско «Лысая певица» звучит эпатажно.
Однако ген, предопределяющий раннее облысение локализован аутосомно, и лысость нельзя считать сцепленной с полом. Ген алопеции— аутосомно-доминантный, причем его экспрессия зависит от уровня андрогенов, производство которых контролируется другими генами.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/