


102 4 Renal Tumors in Children
– Neuroepithelial tumors of the kidney
Age at Presentation for Renal Malignancies |
– |
Lymphoma |
|
|
|||
1. |
Wilms tumor |
|
|
– |
Renal medullary carcinoma |
|
|
|
• Unilateral Wilms tumor: 1–11 year |
– Desmoplastic small round cell tumor of the |
|||||
|
(Mean 3.5 year) |
|
|
kidney |
|
|
|
|
• Bilateral |
Wilms |
tumor: |
– |
Others |
(Fibroma, |
Leiomyoma, |
|
2 months–2 year (Mean 15 months) |
|
Leiomyosarcoma, Primary |
renal synovial |
|||
2. |
Nephroblastomatosis: 6–18 months |
|
sarcoma, Anaplastic sarcoma of the kidney) |
||||
3.Renal cell carcinoma: 6 months–60 year (Commonly 10–20 year)
4. |
Mesoblastic |
nephroma: |
0–1 |
year |
4.2 |
Wilms’ Tumor |
||
|
(Commonly 1–3 months) |
|
|
|
|
|||
5. |
Multilocular cystic renal tumor |
|
4.2.1 |
Introduction |
||||
|
• Cystic nephroma: Adult female |
• Wilms’ (/vɪlmz/) tumor, or nephroblastoma is |
||||||
|
• |
Cystic |
|
partially differentiated |
||||
|
|
nephroblastoma: 3 months–4 year |
one of the commonest tumors of the kidneys |
|||||
|
|
(Commonly 1–2 year) |
|
|
that typically occurs in children. |
|||
6. |
Clear cell sarcoma: 1–4 year (Mean |
• It is named after Dr. Max Wilms, the German |
||||||
|
2 year) |
|
|
|
|
surgeon (1867–1918) who first described it. |
||
7. |
Rhabdoid tumor: 6 months–9 year |
• Wilms’ tumor, or nephroblastoma is a malig- |
||||||
|
(Commonly 6–12 months) |
|
nant tumor of the kidneys that typically occurs |
|||||
8. |
Angiomyolipoma: 6–41 |
year |
(Mean |
in children (Fig. 4.1). |
||||
|
10 year) |
|
|
|
|
• It makes up 87 % of pediatric renal tumors. |
||
9. |
Renal |
medullary |
carcinoma: |
• Wilms tumor is the fourth most common |
||||
|
10–39 year (Mean 20 year) |
|
childhood cancer. |
|||||
10. |
Ossifying |
renal tumor |
of infancy: |
• It is also the most common abdominal malig- |
||||
|
6 |
days–14 |
months |
(Commonly |
nancy in children. |
|||
|
1–3 months) |
|
|
|
• Wilms’ tumor accounts for 6–7 % of all child- |
|||
11. |
Metanephric |
adenoma: |
15 months– |
hood cancers. |
||||
|
83 year |
|
|
|
|
• In North America about 450–500 new cases of |
||
12. |
Lymphoma |
|
|
|
|
Wilms tumor are diagnosed each year. |
||
•Hodgkin lymphoma: >10 year (Commonly late teens
•Non-Hodgkin lymphoma: Any age
(Commonly child <10 year)
WILMS
TUMOR
–Wilms’ tumor
–Cystic partially differentiated nephroblastoma
– |
Mesoblastic nephroma |
|
|
|
NORMAL |
|
URETER |
||||||
– |
Renal cell carcinoma |
|
|
KIDNEYY |
||
|
|
|||||
|
|
|
||||
|
|
|
|
–Clear cell sarcoma
–Rhabdoid tumor of the kidney
– |
Ossifying renal tumor of infancy |
Fig. 4.1 A clinical photograph showing Wilms tumor |
|
– |
Angiomyolipoma |
||
arising from the upper part of the kidney |
4.2 Wilms’ Tumor |
103 |
|
|
•Wilms tumor is relatively more common in blacks than in whites and is rare in East Asians.
•For unknown reasons, Wilms tumor is more common among children of African descent.
•Most cases of Wilms’ tumor occur among children between 3 and 3.5 years old.
•Girls are slightly more likely to develop Wilms’ tumor than are boys.
•The majority (75 %) of Wilms’ tumor cases occur in otherwise normal children.
•In about 25 % of cases, Wilms’ tumors are associated with other developmental abnormalities.
•Most nephroblastomas are unilateral affecting one kidney only.
•This however is not the case in patients with Denys-Drash syndrome who mostly have bilateral or multiple tumors.
•The majority of nephroblastomas are unilateral and bilateral Wilms tumor are seen in 5–10 % of cases.
•In 5–10 % of patients, both kidneys are affected:
–At the same time (synchronous bilateral Wilms tumor).
–Or one after the other (metachronous bilateral Wilms tumor).
•Wilms’ tumor has a classic histologic picture which is called triphasic composed of epithelial, blastemal, and stromal elements.
•Approximately 80–90 % of Wilms’ tumors have favorable histology.
•About 3–7 % of Wilms’ tumors are characterized by anaplastic changes. If these changes are present diffusely throughout the tumor (diffuse anaplasia), they are predictive of a poor outcome.
•Wilms’ tumors are usually encapsulated and vascularized and in cases of metastasis it is usually to the lungs.
•Two renal tumor types (Clear cell sarcoma of the kidney and Rhabdoid tumor of the kidney) were previously included in the category with unfavorable Wilms’ tumors. Currently, these are considered separate malignant entities.
•The overall prognosis of Wilms’ tumor following surgical excision is excellent and the
survival of these patients has improved markedly over the years.
• At present, with early diagnosis and current multimodality therapy, approximately 80–90 % of children with Wilms tumor survive.
–This is attributed to The National Wilms’ Tumor Study Group (NWTSG) and the International Society of Pediatric Oncology (SIOP) who have identified several chemotherapeutic agents through their clinical trials.
•Wilms tumor arises from persistent metanephric blastemal.
•Pathologically, Wilms tumors are described as being triphasic made up of three elements:
–Metanephric blastema
–Stroma (mesenchyme)
–Epithelium
•Add to this the presence of abortive tubules and glomeruli surrounded by a spindled cell stroma. The stroma may include striated muscle, cartilage, bone, fat tissue, and fibrous tissue.
•The mesenchymal component may include cells showing rhabdomyoid differentiation or malignancy (rhabdomyosarcomatous Wilms).
•Pathologically, Wilms tumors is divided into two prognostic groups:
–Favorable: The tumor contains well developed components.
–Anaplastic: The tumor contains diffuse anaplasia (poorly developed cells).
•It was shown that mutations of the WT1 gene on chromosome 11p13 are seen in approximately 20 % of Wilms tumors.
•At least half of the Wilms tumors with mutations in WT1 also carry mutations in CTNNB1, the gene encoding the proto-oncogene beta-catenin.
•It was shown also that a gene on the X chromosome, WTX, is inactivated in up to 30 % of Wilms tumor cases.
•A tumor biopsy is not typically performed due to the risk of creating fragments of cancer tissue and seeding the abdomen with malignant cells.
•Children with Beckwith-Wiedemann syndrome, WAGR syndrome and Denys-Drash
104 |
4 Renal Tumors in Children |
|
|
syndrome, have increased risk of Wilms tumor.
•There is a definite genetic predisposition to Wilms’ Tumor in children with aniridia. This is due to deletions in the p13 band on chromosome 11.
•The treatment of unilateral Wilms’ tumor is nephrectomy followed by chemotherapy, with or without postoperative radiotherapy. Chemotherapy regimens typically comprise vincristine and dactinomycin; doxorubicin and then cyclophosphamide and etoposide are added for increasingly high-risk disease.
•Wilms tumor may spread to the lungs, liver, or nearby lymph nodes.
•Wilms tumor is known to be associated with congenital anomalies:
–Cryptorchidism (2.8 %)
–Hemihypertrophy (2.5 %)
–Hypospadias (1.8 %)
–Sporadic aniridia
•Two loci on chromosome 11 have been implicated in the genesis of Wilms tumors.
–Locus 11p13 is known as the WT1gene
–Locus 11p15 is known as the WT2 gene.
–An abnormal WT1 gene is present in patients with WAGR syndrome:
•Wilms tumor
•Aniridia
•Genitourinary abnormalities
•Mental retardation
–An abnormal WT1 gene is present in patients with Denys-Drash syndrome:
•Male pseudohermaphroditism
•Progressive glomerulonephritis
–An abnormal WT2 gene is present in patients with Beckwith-Wiedemann syndrome or hemihypertrophy.
–The genetics of Wilms tumor appear to be multifactorial, and abnormalities at other sites, including chromosomes 1, 12, and 8, are also recognized.
–Familial Wilms tumor is rare, occurring in approximately 1 % of cases and is not associated with mutations in chromosome 11.
•Screening for Wilms tumor in patients with associated syndromes should begin at 6 months of age with initial computed tomography (CT)
followed by serial ultrasonography (US) every 3 months up to 7 years of age because the risk of developing Wilms tumor after the age of 7 years decreases significantly.
•Wilms tumor occasionally arises in the extrarenal retroperitoneum, presumably within mesonephric remnants (Extra Renal Wilms Tumor).
•The “teratoid Wilms tumor”:
–This is a Wilms tumor characterized by tissue differentiation within the tumor cells such as bone, cartilage, and muscle.
•The vast majority of Wilms tumors demonstrate favorable rather than unfavorable histopathologic findings and anaplasia correlates directly with a negative prognosis and resistance to chemotherapy. It is characterized by atypical mitoses or hyperchromatic cells with large nuclei.
4.2.2Etiology
•Wilms tumor is thought to be caused by alterations of genes responsible for normal genitourinary development.
•This is supported by the common congenital urological anomalies associated with Wilms tumor including:
–Cryptorchidism
–A double collecting system
–Horseshoe kidney
–Hypospadias
•WT1 gene and other genetic loci
–WT1 encodes a transcription factor critical to normal renal and gonadal development.
–WT1, the first Wilms tumor suppressor gene at chromosomal band 11p13, was identified in children with Wilms tumor who also had aniridia, genitourinary anomalies, and mental retardation (WAGR syndrome).
–Karyotypic analysis revealed constitutional deletions within the short arm of 1 copy of chromosome 11.
–The 11p13 locus was subsequently demonstrated to encompass numerous contiguous genes, including the aniridia gene PAX6
4.2 Wilms’ Tumor |
105 |
|
|
and the Wilms tumor suppressor gene WT1.
–A second gene that predisposes individuals to develop Wilms tumor has been identified at chromosomal band 11p15.
–This locus was proposed on the basis of studies in patients with both Wilms tumor and Beckwith-Wiedemann syndrome (BWS).
–Loci at 16q, 1p, 7p, and 17p have also been implicated in the biology of Wilms tumor, although these loci do not seem to predispose individuals to develop a Wilms tumor.
•Beckwith-Wiedemann syndrome (BWS):
–This is an overgrowth syndrome characterized by visceromegaly, macroglossia, and hyperinsulinemic hypoglycemia.
–Patients with BWS are predisposed to have several embryonal neoplasms, including Wilms tumor.
–Several loci for Wilms tumor and BWS have been proposed. These loci include:
•The insulinlike growth factor II gene (IGFII)
•H19
•p57kip2
4.2.3Histopathology
•Wilms tumor arise as a solid intrarenal mass with a pseudocapsule.
•The tumor may also arise from one pole of the kidney.
•The tumor compresses the normal renal parenchyma and leads to distortion of the collecting system.
•Wilms tumor typically spreads by direct extension causing displacement but no encasement of tissues.
•There may be vascular invasion of the renal vein and inferior vena cava with occasional extension into the right atrium.
•Metastases are most commonly found in the lungs (85 % of cases), liver, and regional lymph nodes, and metastatic disease may also produce vascular invasion.
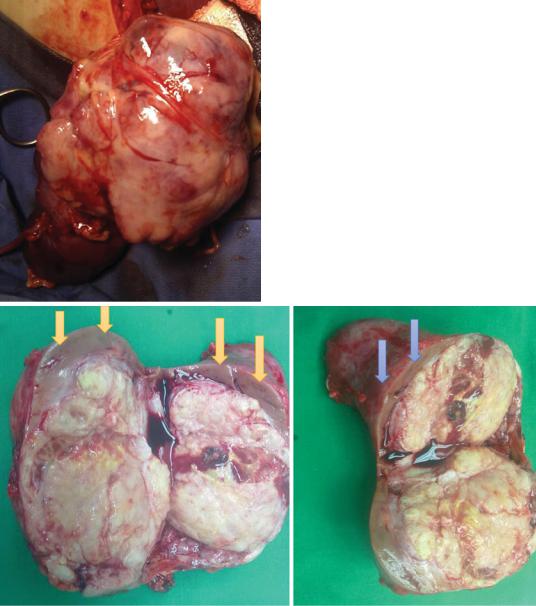
•Pathologically, Wilms’ tumor (Figs. 4.2, 4.3, 4.4, 4.5, 4.6, and 4.7):
–Is usually a large, solitary and encapsulated tumor.
–It compresses the remaining normal kidney parenchyma.
–On cut section, Wilms’ tumor is soft, homogenous and Tan-gray in color and may contain areas of hemorrhage and necrosis.
•Histologically, Wilms’ tumor is a malignant tumor composed of three elements (a triphasic nephroblastoma) (Figs. 4.8, 4.9, 4.10, 4.11, and 4.12):
–Metanephric blastema
–Mesenchymal stroma
–Epithelium
•One of the characteristic histological features of Wilms’ tumor is the presence of abortive tubules and glomeruli surrounded by a spindled cell stroma.
•The stroma may include striated muscles, cartilage, bone, fat tissue, and fibrous tissue.
•Rhabdomyosarcomatous Wilms:
–The mesenchymal stroma may also include cells showing rhabdomyoid differentiation. The rhabdomyoid component may itself show features of malignancy. When this feature is present, it is called rhabdomyosarcomatous Wilms’ tumor. This particular sub-type shows poor response to chemotherapy.
•Wilms’ tumors may be separated into two prognostic groups based on pathologic characteristics:
–Favorable: This contains well developed components.
–Anaplastic (unfavorable): This contains anaplastic cells which could be focal or diffuse. This is associated with higher frequencies of relapse and death especially those with diffuse anaplasia.
•The anaplasia in Wilms’ tumor is classified into two types depending on the extent:
–Diffuse anaplasia
–Localized anaplasia
•Approximately 90 % of all Wilms tumors have favorable histology.
•About 3–7 % of Wilms tumors are characterized by anaplastic changes.
•The presence of diffuse anaplasia throughout the tumor is a predictive of poor outcome.
106 |
4 Renal Tumors in Children |
|
|
Figs. 4.2, 4.3, and 4.4 Clinical photographs showing Wilms tumor. Note the compressed renal tissue at the periphery
•Clear cell sarcoma of the kidney and rhabdoid tumor of the kidney were included in the unfavorable histology in the past and currently these are considered as separate malignant tumors.
•The presence of nephrogenic rests, and dysplastic lesions of metanephric origin, are now believed to represent precursor lesions of Wilms tumor. These lesions are observed in
approximately one third of kidneys affected by Wilms tumors.
•It was also shown that children younger than age 12 months diagnosed with perilobar nephrogenic rests have a markedly increased risk of developing a contralateral Wilms tumor.
•The National Wilms Tumor Study (NWTS) and Societe Internationale D’Oncologie Pediatrique
4.2 Wilms’ Tumor |
107 |
|
|
Fig. 4.5 A clinical photograph showing an excised Wilms tumor. Note the fleshy, homogenous and Tan-gray in color tumor
Fig. 4.7 A clinical photograph showing an excised Wilms tumor. Note the areas of hemorrhage within the tumor
Fig. 4.8 A histological photograph of Wilms’ tumor showing blastema ( ) tubules (
) tubules ( ) and spindle cells
) and spindle cells
Fig. 4.6 A photograph of a resected Wilms’ tumor. Note ( |
) |
the gross appearance of Wilms’ tumor with the normal |
|
part of the kidney being compressed by the tumor |
|
(SIOP) have made large contributions to the modern multimodal treatment, which consists of surgical excision, radiotherapy, and chemotherapy (adjuvant and/or neoadjuvant). These oncologic treatments achieve a remarkable long-term overall survival rate of 90%.
•Most cases of Wilms tumor do not have mutations in any of the genes.
•A gene on the X chromosome, WTX, is inactivated in up to 30 % of Wilms’ tumor cases.
•The gene WT1:
–This is also called Wilms tumor suppressor gene.
–It has been found to make a protein that is found mostly in the fetal kidney and in tissues that give rise to the genitourinary system.
–Inactivation of this gene may be responsible for the occurrence of Wilms tumor.
–Mutations of the WT1 gene on chromosome 11 p 13 are observed in approximately 20 % of Wilms’ tumors.
–At least half of the Wilms’ tumors with mutations in WT1 also carry mutations in CTNNB1, the gene encoding the protooncogene beta-catenin.