

metodichka_Lipidy_2020
.pdfРис. 5-2. Интеграционное взаимодействие липопротеинов в сосудистом русле
- ферментативная деградация зрелых («плазменных») ЛП в «ремнантные», т.е., в которых в кровотоке произошли изменения в количественном и качественном составе липидных компонентов, что привело к формированию иной конформации апобелков. На их поверхности формируются лиганды узнавания, которые обеспечивают направленный транспорт ЛП к соответствующим тканям-мишеням;
-рецептор-опосредованный эндоцитоз (поглощение) ремнантных ЛП, путем образования комплекса с рецептором, сокращения клатрина и инвагинации мембраны, формирования эноцитозных везикул;
-утилизация компонентов ЛП клеткой за счет слияния везикул с лизосомами (образование фагосом) и последующим гидролизом ЛП ферментами.
Для ХМ характерно формирование насцетных ХМ в энтероцитах, где синтезированный апо В-48 связывает ресинтезировавшийся экзогенный ТАГ, образуя белок-липидный комплекс, который покрывается монослоем фосфолипидов.
Насцетные ЛПОНП образуются в гепатоцитах, где синтезируется апоВ-100. Он имеет 5 доменов. Два домена являются ТАГ-связывающими и формируют гидрофобное ядро из ТАГ, куда погружается домен узнавания (связывания с рецепторами). Есть домен, связывающий ЭХС. В процессе созревания насцетные ХМ и ЛПОНП получают от ЛПВП динамические апобелки: апоС-ІІ, апоЕ. Зрелые ХМ и ЛПОНП транспортируются к тканям, где на поверхности капилляров подвергаются гидролизу гепаринзависимой ЛП-липазой (ЛПЛ), которая активируется коферментным апоС-II. ЛПЛ гидролизует ТАГ гидрофобного ядра ХМ в α- положении, расщепляя их на ЖК и ДАГ. ЖК поглощаются клетками тканей и органов, ХМ и ЛПОНП деградируют. Они отдают часть ДАГ, апоС-II в ЛПВП, обогащаясь от них ЭХС. Этот процесс называет гетерообмен. В результате ХМ и ЛПОНП превращаются в «ремнантные» ХМ и ЛППП. У них с апоЕ сформирован лиганд узнавания, соответственно, к апоВ-48/Е и апоВ/Е-рецепторам гепатоцитов, что обеспечивает их эндоцитоз. Часть ЛППП, получив от ЛПВП апоС-ІІІ, подвергаясь затем действию триглицеридлипазы печени (глицеролгидролазы) (ТГЛ), превращается в ЛПНП. Клетки печени, половых желез и надпочечников содержат большое количество рецепторов к ЛПНП (рецепторы апоВ/Е), способны к активному потреблению ХС. Наследственный дефект их синтеза – причина семейной гиперхолестеринемии. Существует три пула ЛПВП – печеночный, кишечный и плазменный. Образование ЛПВП реализуется по нескольким путям, т.ч. это синтез насцетных ЛПВП печенью и энтероцитами (бислойных дисков из апопротеинов и ФЛ), либо взаимодействие новообразованных апопротеинов с мембранами клеток. Затем диффузия свободного ХС из клеток эндотелия, эритроцитов и пр., абсорбция продуктов липолиза ЛПОНП и ХМ (белков – апоА-I, группы С; фосфолипидов, ХС). Взаимодействие дискоидальных насцетных ЛПВП с ЛХАТ приводит к погружению нейтральных ЭХС в ядро частицы и формированию зрелых (сферических) ЛПВП3. Важной особенностью дальнейшей трансформации ЛПВП3 в ЛПВП2 является перенос ЭХС из ЛПВП3 в более крупные ЛП, богатые ТАГ. Белок, переносящий ЭХС, реализует обмен этерифицированного ХС из ЛПВП на ТАГ липопротеинов, содержащих апоВ. Снижение ЭХС и нарастание уровня ТАГ ведет к увеличению размеров и снижению плотности ЛПВП, т.е к превращению ЛПВП3 в ЛПВП2. В

дальнейшем последние претерпят действие ТГЛ синусоидов печени и вновь превращаются в ЛПВП3, возвращаясь в кровоток. Часть ЛПВП3 подвергаются эндоцитозу клетками печени, взаимодействуя со специфическими к ЛПВП апоА-I/Е- рецепторами.
3.Схема обмена липопротеинов в организме.
Липиды пищи агрегируются в хиломикроны. Большая часть содержащихся в них триглицеридов высвобождается в жировую и сышечную ткани в капиллярах. Остатки хиломикронов (содержащие главным образом белок и холестерин) захватываются гепатоцитами.
Эндогенные липиды и холестерин из печени доставляются в жировую и мышечную ткани в виде ЛПОНП. Выход липидов из ЛПОНП и обмен апопротеинами с ЛПВП постепенно превращает ЛПОНП в ЛПНП, которые доставляют холестерин во внепеченочные ткани и возвращают его в печень. Избыточный во внепеченочных тканях холестерин транспортируется обратно в печень в виде ЛПНП, где частично превращается в соли желчных кислот.
Печень захватывает остатки ЛПОНП, ЛПНП и остатки хиломикронов путем опосредованного рецепторами эндоцитоза.
Рис. 5-3. Липиды и липидный транспорт.
4.Нарушения обмена липопротеинов: дис- и гиполипопротеинемии.
Все изменения содержания ЛП в плазме крови, характеризующиеся их повышением, снижением или полным отсутствием, объединяют под названием дислипопротеинемий. Дислипопротеинемия может быть либо специфическим первичным проявлением нарушений в обмене липидов и липопротеинов, либо сопутствующим синдромом при некоторых заболеваниях внутренних органов (вторичные дислипопротеинемии). При успешном лечении основного заболевания они исчезают.
Кгиполипопротеинемиям относят следующие состояния.
1.Абеталипопротеинемия возникает при редком наследственном заболевании – дефекте гена апопротеина В, когда нарушается синтез белков апоВ-100 в печени
иапоВ-48 в кишечнике. В результате в клетках слизистой оболочки кишечника не формируются ХМ, а в печени – ЛПОНП, и в клетках этих органов накапливаются капельки жира.
2.Семейная гипобеталипопротеинемия: концентрация ЛП, содержащих апоВ составляет лишь 10-15% нормального уровня, но организм способен образовывать ХМ.
3.Семейная недостаточность -ЛП (болезнь Тангира): в плазме крови практически не обнаруживаются ЛПВП, а в тканях накапливается большое количество эфиров ХС, у пациентов отсутствует апоС-II, являющийся активатором ЛПЛ, что ведет к характерному для данного состояния повышению концентрации ТАГ в плазме крови.
5.Классификация гиперлипопротеинемий.
Среди гиперлипопротеинемий различают следующие типы.
Тип I - гиперхиломикронемия. Скорость удаления ХМ из кровотока зависит от активности ЛПЛ, присутствия ЛПВП, поставляющих апопротеины С-II и Е для ХМ, активности переноса апоС-II и апоЕ на ХМ. Генетическе дефекты любого из белков, участвующих в метаболизме ХМ, приводят к развитию семейной гиперхиломикронемии – накоплению ХМ в крови. Заболевание проявляется в раннем детстве, характеризуется гепатоспленомегалией, панкреатитом, абдоминальными болями. Как вторичный признак наблюдается у больных сахарным диабетом, нефротическим синдромом, гипотиреозом, а также при злоупотреблении
алкоголем. Лечение: диета с низким содержанием липидов (до 30 г/сут) и высоким содержанием углеводов.
Тип II – семейная гиперхолестеролемия (гипер- -липопротеинемия). Этот тип делят на 2 подтипа: IIа, характеризующийся высоким содержанием в крови ЛПНП, и IIб – с повышенным уровнем как ЛПНП, так и ЛПОНП. Заболевание связано с нарушением рецепции и катаболизма ЛПНП (дефект клеточных рецепторов для ЛПНП или изменение структуры ЛПНП), сопровождается усилением биосинтеза холестерола, апо-В и ЛПНП. Это наиболее серьезная патология в обмене ЛП: степень риска развития ИБС у пациентов с этим типом нарушения возрастает в 1020 раз по сравнению со здоровыми лицами. Как вторичное явление гиперлипопротеинемия II типа может развиваться при гипотиреозе, нефротическом синдроме. Лечение: диета с низким содержанием холестерола и насыщенных жиров.
Тип III – дис- -липопротеинемия (широкополосная беталипопротенемия)
обусловлена аномальным составом ЛПОНП. Они обогащены свободным ХС и дефектным апо-Е, тормозящим активность печеночной ТАГ-липазы. Это ведет к нарушениям катаболизма ХМ и ЛПОНП. Заболевание проявляется в возрасте 30-50 лет. Состояние характерируется высоким содержанием остатков ЛПОНП, гиперхолестеролемией и триацилглицеролемией, наблюдаются ксантомы, атеросклеротические поражения периферических и коронарных сосудов. Лечение: диетотерапия, направленная на снижение веса.
Тип IV – гиперпре-b-липопротеинемия (гипертриацилглицеролемия). Первичный вариант обусловлен уменьшением активности ЛПЛ, повышение уровня ТАГ в плазме крови происходит за счет фракции ЛПОНП, аккумуляции ХМ при этом не наблюдается. Встречается только у взрослых, характеризуется развитием атеросклероза сначала коронарных, затем периферических артерий. Заболевание часто сопровождается понижением толерантности к глюкозе. Как вторичное проявление встречается при панкреатите, алкоголизме. Лечение: диетотерапия, направленная на снижение веса.
Тип V – гиперпре-b-липопротеинемия с гиперхиломикронемией. При этом типе патологии изменения фракций ЛП крови носят сложный характер: повышено
содержание ХМ и ЛПОНП, выраженность фракций ЛПНП и ЛПВП уменьшена. Больные часто имеют избыточную массу тела, возможно развитие гепатоспленомегалии, панкреатита, атеросклероз развивается не во всех случаях. Как вторичное явление гиперлипопротеинемия V типа может наблюдаться при инсулинзависимом сахарном диабете, гипотиреозе, панкреатите, алкоголизме, гликогенозе I типа. Лечение: диетотерапия, направленная на снижение веса, диета с невысоким содержанием углеводов и жиров.
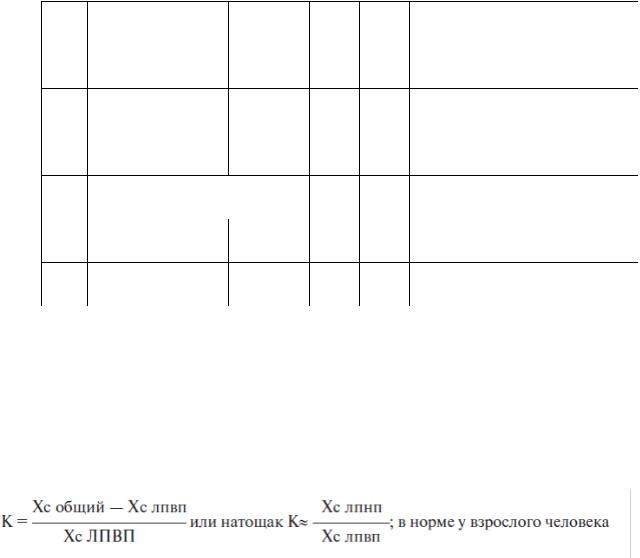
Табл. 2
Классификация гиперлипопротеинемий, принятая ВОЗ
Тип |
ХМ |
ЛПОНП |
ЛПНП |
ХС |
ТАГ |
Нарушения |
I |
↑ |
N |
N |
N |
↑↑ |
↑ХМ |
IIa |
- |
N |
↑↑ |
↑↑ |
N |
↑ЛПНП |
IIb |
- |
↑ |
↑ |
↑ |
↑ |
↑ЛПНП и ЛПОНП |
III |
- |
Флотирующие β-ЛП |
↑ |
↑ |
↑ЛППП и ремнантных ХМ |
|
IV |
- |
↑ |
N |
N(↑) |
↑ |
↑ЛПОНП |
V |
↑ |
↑ |
N |
N(↑) |
↑↑ |
↑ХМ и ЛПОНП |
6. Гиперхолестеринемия, ее причины, последствия.
Концентрация холестерола в крови взрослых людей составляет 200±50 мг/дл (5,2±1,2 ммоль/л) и, как правило, увеличивается с возрастом. Превышение нормальной концентрации холестерола в крови называют гиперхолестеролемией.
Этот показатель не должен превышать 3,5.
Гиперхолестеролемия является одной из форм гиперлипопротеинемии. Это нарушение липидного обмена сопровождается повышением в крови фракции атерогенных ЛПНП, которые содержат более 50% холестерола и его эфиров. Гиперхолестеролемия может привести к развитию атеросклероза.
Причины гиперхолестеролемии Семейная гиперхолестеролемия вызвана мутациями в гене апоВ-100-
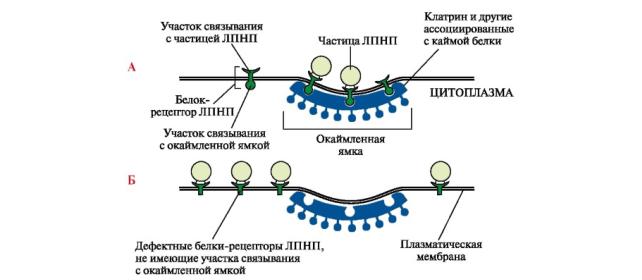
рецептора. Мутации в разных участках этого гена могут приводить к снижению количества апоВ-100-рецепторов в плазматической мембране клеток или неправильному положению в мембране (вне окаймленной ямки) и нарушению эндоцитоза (рис. 5-1). Повышаются содержание ЛПНП и время их циркуляции в крови, концентрация холестерола у больных может превышать норму в 4 раза (норма 200±50 мг/дл). Время полураспада ЛПНП возрастает до 4-6 сут (норма 21/2 сут), что способствует образованию измененных, модифицированных форм ЛПНП, обладающих высокой атерогенностью.
Рис. 5-4. Положение рецептора ЛПНП в норме и при нарушении его структуры
А. Положение апоВ-100-рецепторов в окаймленной ямке. Б. Положение апоБ-100- рецепторов вне окаймленной ямки
Наследственные дефекты апоВ-100. Нарушения структуры этого апобелка снижает комплементарность апоВ-100 к апоВ-100-рецептору и поступления ЛПНП в клетки. Повышение содержания ЛПНП в крови приводит к гиперхолестеролемии.
Вторичные гиперхолестеролемии. Причиной гиперхолестеролемии может стать гиперкалорийное питание, вызванное избыточным содержанием в рационе углеводов и липидов, так как все субстраты для синтеза холестерола - ацетил-КоА, NADPH, АТФ - образуются из углеводов. Избыточный синтез ТАГ и холестерола в печени вызывает повышенное формирование ЛПОНП, которые в крови превращаются в ЛПНП. Но поглощение липопротеинов клетками тканей зависит не
от количества ЛПНП в крови, а от количества рецепторов в плазматических мембранах, которое в свою очередь регулируется холестеролом. В крови повышается содержание ЛПНП, а следовательно, и холестерола.
Вторичные гиперхолестеролемии могут быть вызваны химическими модификациями белков и липидов ЛПНП.
Типы модификаций:
•гликозилирование апоВ-100 в составе ЛПНП или апоВ-100-рецепторов. В норме у здоровых людей с очень небольшой скоростью протекает неферментативное гликозилирование этих белков. Остатки глюкозы присоединяются к свободным H2N- группам белков, изменяется их конформация. Модифицированные ЛПНП* не узнаются рецепторами и остаются в крови. У больных сахарным диабетом этот процесс протекает очень активно.
•перекисное окисление липидов ЛПНП: ПОЛ подвергаются жирные кислоты в составе липидов липопротеинов. Постоянно возникающие в организме свободные радикалы активируют образование гидроперекисей полиненасыщенных жирных кислот фосфолипидов, ТАГ и эфиров холестерола. Образовавшиеся гидроперекиси могут реагировать с Н2N-группами апоВ-100. При этом изменяются заряд апобелка, конформация и сродство к апоВ-100-рецепторам.
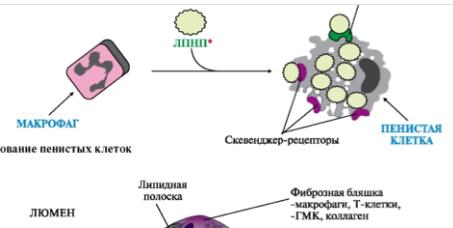
Возрастают время циркуляции ЛПНП* в крови и степень их повреждения, поэтому снижается вероятность их взаимодействия с апоВ-100-рецепторами. Эти частицы как чужеродные поглощаются макрофагами с помощью скевенджеррецепторов (R-мусорщиков). Количество скевенджер-рецепторов не регулируется холестеролом, поэтому макрофаги захватывают очень много модифицированных ЛПНП*, придающих этим клеткам характерный пенистый вид (рис. 5-2).
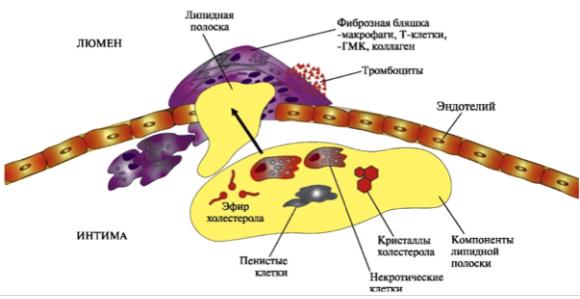
Рис. 5-5. Образование пенистых клеток.
7. Механизм формирования атеросклеросклеротической бляшки.
Под влиянием эндогенных факторов ЛПНП и ЛПОНП, способные проникать в интиму сосудов, подвергаются трансформации. Модифицированные ЛП поглощаются макрофагами. Этот процесс не регулируется количеством поглощенного ХС, как в случае его поступления в клетки через специфические рецепторы, поэтому макрофаги перегружаются ХС и превращаются в «пенистые клетки», которые проникают в субэндотелиальное пространство и вызывают его повреждение. В область дефекта устремляются тромбоциты, которые с помощью регуляторных факторов стимулируют увеличение количества и активности гладкомышечных клеток.
Рис. 5-6. Атеросклеротическая бляшка
Это приводит к формированию липидных пятен или полосок в стенке кровеносных сосудов. На этой стадии эндотелий сосудов может сохранять свою структуру. При увеличении количества пенистых клеток происходит повреждение эндотелия и активация тромбоцитов. В результате они секретируют тромбоксан, который стимулирует агрегацию тромбоцитов, а также начинают продуцировать тромбоцитарный фактор роста, стимулирующий пролиферацию гладкомышечных клеток. Последние мигрируют из медиального во внутренний слой (интиму) артериальной стенки, способствуя, таким образом, росту бляшки. Далее происходит прорастание
бляшки фиброзной тканью, клетки под фиброзной оболочкой некротизируются, а ХС откладывается в межклеточном пространстве. На последних стадиях развития бляшка пропитывается солями кальция и становится очень плотной. В области бляшки часто образуются тромбы, перекрывающие просвет сосуда, что приводит к острому нарушению кровообращения в соответствующем участке ткани и развитию инфаркта.
Следовательно, механизм развития атеросклеротической бляшки заключается
вследующем:
•повышается содержание ЛПНП в крови;
•повышается время жизни ЛПНП;
•повышается содержание в крови поврежденных в результате ПОЛ и гликозилирования ЛПНП*;
•понижается поглощение ЛПНП* клетками тканей;
•повышается поглощение ЛПНП* макрофагами с помощью скевенджеррецепторов;
•перегруженные холестеролом макрофаги превращаются в «пенистые» клетки;
•«пенистые» клетки проникают под слой эндотелиальных клеток;
•повышение количества «пенистых» клеток вызывает повреждение эндотелия.
На поврежденной поверхности происходит агрегация тромбоцитов и секреция тромбоксанов ТХА2, которые: ↑ агрегацию тромбоцитов и вызывают сокращение стенки сосуда.
Тромбоцитарного фактора роста, который:
•↑ деление (пролиферацию) ГМК и их миграцию в область повреждения;
•↑ секрецию ГМК - коллагена, эластина;
•↑ рост бляшки
•↓ просвет сосуда;
•↑ вероятность образования тромба в области бляшки.
Внутри атеросклеротической бляшки клетки погибают. Разрыв оболочки бляшки вызывает кровотечение, происходит быстрое образование тромба, закрывающего