

29. Сахарный диабет инсулинзависимый (ИЗСД, I тип): биохимическая диагностика, механизмы развития метаболических нарушений (гипергликемия, холестеринемия, кетонемия, ацидоз, гликозилирование белков), биохимические особенности детского возраста
САХАРНЫЙ ДИАБЕТ I типа
СД I типа — заболевание, которое возникает вследствие абсолютного дефицита инсули-на, вызванного аутоиммунным разрушением β-клеток поджелудочной железы. СД I типа по-ражает в большинстве случаев детей, подростков и молодых людей до 30 лет, но может про-явиться в любом возрасте. СД I типа редко является семейным заболеванием (10-15% всех случаев).
Причины СД I типа
1.Генетическая предрасположенность. Генетические дефекты ведущие к СД могут реа-лизоваться в клетках иммунной системы и β-клетках поджелудочной железы. В β-клетках известно около 20 генов, способствующих развитию СД I типа. В 60-70% случа-ях СД I типа связан с наличием в 6 хромосоме HLA региона генов DR3, DR4 и DQ.
2.Действие на β-клетки β-цитотропных вирусов (оспа, краснуха, корь, паротит, Кокса-ки, аденовирус, цитомегаловирус), химических и других диабетогенов.
Вариант 1
При наличии генетического дефекта, на поверхности β-клеток накапливаются антиге-ны, имеющие схожую аминокислотную последовательность с β- цитотропными вирусами.
В случае возникновения инфекции β-цитотропных вирусов, развиваются иммунные реакции против этих вирусов и аутоиммунные реакции против схожих антигенов β-клеток. Реакция идет с участием моноцитов, Т- лимфоцитов, антител к β-клеткам, инсулину, глута-мат декарбоксилазе (фермент 64кДа, находиться на мембране β-клеток). В результате аутоиммунные реакции вызывают гибель β-клеток.
Вариант 2
При действии на β-клетки с генотипом HLA β-цитотропных вирусов или диабетогенов на поверхности β-клеток происходит изменение антигенов.
50
На измененные антигены β-клетки развиваются аутоиммунные реакции. Аутоиммунные реакции вызывают гибель β-клеток.
Вариант 3
β-цитотропные вирусы имеют схожую последовательность аминокислот с глутамат декарбоксилазой β-клеток. Генетический дефект СД8+ лимфоцитов (Т-супрессоров) не позволяет им отличить аминокислотную последовательность вируса и глутамат декарбоксилазы, поэтому при возникновении инфекции, Т-лимфоциты реагируют на глутамат декарбоксилазу β-клеток как на вирус.
Вариант 4
Некоторые β-цитотропные вирусы и химические диабетогены, например, производные нитрозомочевины, нитрозамины, аллоксан самостоятельно и избирательно поражают β-клетки, вызывая их лизис;
Стадии развития СД I типа
1.Стадия генетической предрасположенности. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
2.Стадия провоцирующих событий. Инфекция β-цитотропных вирусов или действие химических диабетогенов. Протекает без клинических симптомов;
3.Стадия явных иммунных аномалий. Развитие смешанных аутоиммунных реакций против β-клеток. Ресурсы инсулина достаточны. Протекает без клинических симптомов.
Развивается от 2-3 месяцев до 2-3 лет;
4.Стадия латентного диабета. Гибель 75% β-клеток,
небольшое снижение инсулина, гипергликемия при нагрузочных пробах, снижение аутоиммунных процессов. Протекает без клинических симптомов;
5.Явный диабет. Гибель 80-90% β-клеток, заметное снижение инсулина, гипергликемия натощак, нет или слабые аутоиммунные реакции. Появляются клинические симптомы. Развивается 2 года. Необходима инсулинотерапия;
6.Терминальный диабет. Полная гибель β-клеток, высокая потребность в инсулинотерапии, аутоиммунные проявления
51
снижены или их нет. Выраженные клинические проявления, появляются ангиопатии. Развивается до 3,5 лет;
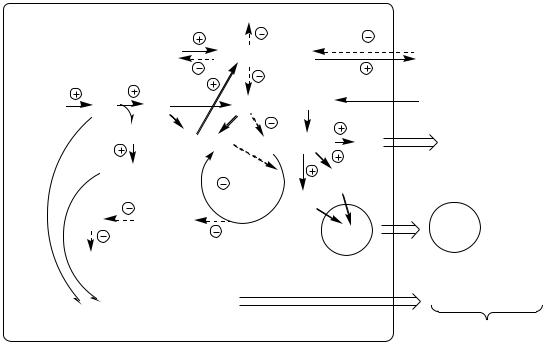
Изменения метаболизма при СД I типа
При СД I типа исчезает инсулин, т.к. инсулин ингибитор секреции глюкагона, в крови происходит увеличение глюкагона.
Изменения в углеводном обмене
Ï Å× ÅÍ Ü |
|
|
Ï ÔØ |
|
ÊÐÎ ÂÜ |
|
|
|
|
|
|
ãëè êî ãåí |
|
Ãëþ êî çà |
|
Ãëþ êî çà |
|
Белки ÀÊ |
ÀÊ |
|
Ï ÂÊ |
ÑÆÊ |
ÑÆÊ |
|
|
|
|||
|
NH3 |
Ù ÓÊ |
АцетилКо А ÊÒ |
ÊÒ |
|
|
|
||||
м о чевин а |
|
ÖÒÊ |
ÕÑ |
|
|
|
|
|
|
||
|
|
|
|
|
|
ÄÖ |
Í ÀÄÍ 2 |
|
ÒÃ |
|
|
|
|
ÕÑ |
|||
|
|
|
|
|
ÒÃ |
ÀÒÔ |
|
|
|
ËÏ Î Í Ï |
ËÏ Î Í Ï |
|
|
|
|
|
|
ÀÊ, ì î ÷åâè í à |
|
|
ÀÊ, ì î ÷åâè í à |
||
Àçî ò
В печени дефицит инсулина и избыток глюкагона стимулирует реакции глюконеогенеза, гликогенолиза и ингибирует реакции гликолиза, ПФШ и синтеза гликогена. В результате в печени глюкозы больше образуется, чем потребляется.
Так как реакции глюконеогенеза протекают через ЩУК, он, образовавшись из ПВК, аспартата и малата, активно вовлекается в глюконеогенез, вместо того чтобы включаться в ЦТК. В результате ЦТК и ДЦ тормозится, снижается образование АТФ, возникает энергодефицит.
В инсулинзависимых тканях (мышцы, жировая ткань) дефицит инсулина препятствует поступлению глюкозы в клетки и ее использованию в реакциях гликолиза, ПФШ и синтеза гликогена. Блокирование ЦТК и ДЦ также вызывает энергодефицит.
Снижение потребления глюкозы инсулинзависимыми тканями и усиление ее образования в печени приводит к гипергликемии. Когда гипергликемия превышает концентрационный почечный порог возникает глюкозурия.
52
Глюкозурия – наличие глюкозы моче. В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу. Если уровень глюкозы превышает в крови 9-10 ммоль/л, глюкоза не успевает полностью реабсорбироваться из первичной мочи и частично выводится с вторичной мочой.
У больных с СД после приёма пищи концентрация глюкозы в крови может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе.
ÀÄÈ Ï Î ÖÈ Ò |
|
|
ÊÐÎ ÂÜ |
|
|
|
|
|
|
|
Ãëþ êî çà |
|
Ãëþ êî çà |
|
ÒÃ |
ÆÊ |
|
ÆÊ |
|
|
глицеро -3Ф |
|
ãë è öåðî -3Ô |
|
|
|
|
|
|
|
|
|
|
Деф ицит ин сулин а |
|
|
ÆÊ |
ËÏ Ë |
ÕÑ |
|
|
|
||
|
|
|
ÒÃ |
|
|
|
|
|
|
|
|
|
|
ËÏ Î Í Ï |
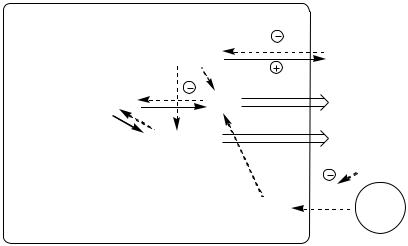
Изменения в липидном обмене
Дефицит АТФ, НАДФН2, инсулина и избыток глюкагона тормозят липогенез и усиливают липолиз в жировой ткани. В результате в крови повышается концентрация свободных жирных кислот, которые поступают в печень и окисляются там до Ацетил-КоА. АцетилКоА при дефиците ЩУК не может включаться в ЦТК. Поэтому он накапливается и поступает на альтернативные пути: синтез кетоновых тел (ацетоуксусная, β-гидроксимасляная кислоты) и холестерина.
Внорме кетоновые тела являются источником энергии для аэробных тканей, они превращаются в АцетилКоА, который окисляется в ЦТК. Так как ЦТК заблокирован дефицитом ЩУК, кетоновые тела накапливаются в крови и вызывают кетонемию. Кетонемия усугубляет недостаточность инсулина, подавляя остаточную секреторную активность β-клеток. Когда кетонемия превышает концентрационный почечный порог (выше 20 мг/дл, иногда до 100 мг/дл) возникает кетонурия. Кетонурия – наличие кетоновых тел в моче.
Втканях ацетоуксусная кислота частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии.
53
Липопротеины крови поставляют субстраты для липогенеза в тканях. Дефицит инсулина блокирует липогенез в жировой ткани, ингибирует липопротеинлипазу в сосудах, это препятствует расщеплению липопротеинов крови (в основном, ЛПОНП), в результате они накапливаются,
вызывая гиперлипопротеинемию.
Изменения в белковом обмене
Энергодефицит, недостаток инсулина и избыток глюкагона приводит к снижению скорости синтеза белков в организме и усилению их распада, что повышает концентрацию аминокислот в крови. Аминокислоты поступают в печень и дезаминируются до кетокислот. Кетокислоты включаются в глюконеогенез, что усиливает гипергликемию. Из аммиака активно синтезируется мочевина. Повышение в крови аммиака, мочевины, аминокислот вызывает азотемию – увеличение концентрации азота в крови. Азотемия приводит к азотурии – увеличению концентрации азота в моче. Развивается отрицательный азотистый баланс. Катаболизм белков ведет к миодистрофии и вторичному иммунодефициту.
Изменения в водно-солевом обмене
Поскольку возможности почек ограничены, высокие концентрации глюкозы, кетоновых тел и мочевины не успевают реабсорбироваться из первичной мочи. Они создают в первичной моче высокое осмотическое давление, которое препятствует реабсорбции воды в кровь и образованию вторичной мочи. У таких пациентов развивается полиурия, выделение мочи возрастает до 3—4 л в сутки (в некоторых случаях до 8—9 л). Потеря воды вызывает постоянную жажду или полидипсию. Без частого питья, полиурия может приводить к обезвоживанию организма. Потеря с мочой глюкозы усугубляет энергодефицит, может увеличить аппетит и полифагию. С первичной мочой из организма уходят некоторые полезные минеральные компоненты, что приводит к нарушению минерального обмена.
Высокие концентрации глюкозы, кетоновых тел и мочевины создают в плазме крови значительное осмотическое давление, которое способствует дегидратации тканей. Кроме воды ткани теряют электролиты, прежде всего ионы К+, Na+, С1-, НСО3-.
Изменение в газообмене тканей
Общая дегидратация организма, вызванная полиурией и дегидратацией тканей приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Причиной
54
гипоксии является также гликозилирование Hb в HbA1c, который не переносит О2 к тканям. Гипоксия ведет к энергодефициту и накоплению в организме
лактата.
Изменения в кислотно-основном равновесии
Накопление кетоновых тел, лактата и потеря щелочных валентностей с мочой снижает буферную ёмкость крови и вызывает ацидоз.
Симптомы СД I типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены. Общая слабость, похудание, снижение трудоспособности, сонливость. Ожирение отсутствует. Повышенный аппетит при кетоацидозе сменяется анорексией. Развивается быстро, склонен к развитию кетоацидотической комы.
30. Сахарный диабет инсулиннезависимый (ИНЗСД, II тип): метаболические нарушения, биохимическая диагностика,
механизмы развития метаболических нарушений (гипергликемия, холестеринемия, липидемия, гликозилирование белков), биохимические особенности детского возраста
САХАРНЫЙ ДИАБЕТ II типа
СД II типа представляет собой группу гетерогенных нарушений углеводного обмена. СД II типа не инсулинозависимый, не склонен к кетоацидотической коме, не имеет антител к β-клеткам, не аутоиммунной природы, не имеет связи с определенными HLA фенотипами. Ожирение в 80%. На долю СД II типа приходится примерно 85-90% всех случаев СД, он поражает людей, как правило, старше 40 лет и характеризуется высокой частотой семейных форм (риск СД II типа у ближайших родственников больного достигает 50%, тогда как при СД I типа он не превышает 10%). СД II типа поражает преимущественно жителей развитых стран, особенно горожан.
Воснове СД II типа лежат множество причин. СД II типа развивается при:
генетических дефектах рецепторов инсулина, у них снижается чувствительность к инсулину;
синтезе дефектного инсулина с низкой биологической активностью (мутация гена инсулина: в позиции 24 В-цепи вместо фен присутствует лей);
55
нарушении превращения проинсулина в инсулин;
нарушении секреции инсулина;
повреждении инсулина и его рецепторов антителами;
повышения скорости катаболизма инсулина;
действия контринсулярных гормонов (создают гипеинсулинемию, которая вызывает инсулинорезистентность);
нарушении глюкозочувствительного механизма  клеток (мутации гена глюкокиназы) и т.д.
клеток (мутации гена глюкокиназы) и т.д.
Основным провоцирующим фактором СД II типа служит ожирение.
Стадии СД II типа
1.Стадия генетической предрасположенности. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
2.Стадия латентного диабета. Гипергликемия при нагрузочных пробах. Протекает без клинических симптомов СД;
3.Явный диабет. Гипергликемия натощак. Появляются клинические симптомы.
Симптомы СД II типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены умеренно или отсутствуют. Часто ожирение (у 80-90% больных).
Изменения метаболизма при СД II типа
Относительный дефицит инсулина вызывает метаболические нарушения, схожие с теми которые возникают при абсолютном дефиците инсулина, однако эти нарушения менее выражены, а у 50% больных с ожирением и умеренной гипергликемией СД II типа вообще протекает бессимптомно.
В отличие от абсолютного дефицита инсулина, при относительном дефиците инсулина, влияние инсулина сохраняется на жировую ткань, имеющую высокое содержание рецепторов к инсулину. Инсулин в жировой ткани стимулирует липогенез, блокирует липолиз и выход жирных кислот в кровь, поэтому при СД II типа не наблюдается кетоацидоз, масса тела не уменьшается, а наоборот развивается ожирение. Таким образом, ожирение,
56
с одной стороны, важнейший фактор риска, а с другой — одно из ранних проявлений СД II типа.
При СД II типа наблюдается гиперинсулинемия (80%), артериальная гипертензия (50%), гиперлипидемия (50%), атеросклероз, нейропатия (15%) и диабетическая нефропатия (5%).
Осложнения СД
Острые осложнения сахарного диабета. Механизмы развития диабетической комы
Острые осложнения специфичны для СД I и II типа.
Дегидратация тканей головного мозга в первую очередь, а также нарушения обмена веществ в нервной ткани могут приводить к развитию острых осложнений в виде коматозных состояний. Кома это крайне тяжелое состояние, характеризующееся глубоким угнетением ЦНС, стойкой потерей сознания, утратой реакций на внешние раздражители любой интенсивности. Коматозные состояния при СД могут проявляться в трёх формах: кетоацидотической, гиперосмолярной и лактоацидотической.
Кетоацидотическая кома возникает при СД I типа, когда концентрация кетоновых тел становится выше 100 мг/дл (до 400-500мг/дл).
Гиперкетонемия приводит к:
1)ацидозу, который блокирует активность большинства ферментов, в первую дыхательных, что вызывает гипоксию и снижение синтеза АТФ.
2)гиперосмолярности, которая приводит к дегидратации тканей и нарушению водно-электролитного равновесия, с потерей ионов калия, натрия, фосфора, магния, кальция, бикарбонатов.
Это при определенной выраженности и вызывает коматозное состояние с падением артериального давления и развитием острой почечной недостаточности.
Возникающая гипокалиемия ведет к гипотонии гладкой и поперечнополосатой мускулатуры, снижению тонуса сосудов, падению АД, сердечной аритмии, гипотонии дыхательной мускулатуры с развитием острой
57
дыхательной недостаточности; атонии ЖКТ с парезом желудка и развитием кишечной непроходимости развивается выраженная гипоксия. В общей причине смертности она занимает 2-4 %.
Гиперосмолярная кома характерна для СД II типа, она наблюдается при высокой гипергликемии. У большинства высокая гипергликемия обусловлена сопутствующим нарушением функции почек, ее провоцируют стресс, травма, резкая дегидратация организма (рвота, диарея, ожоги, кровопотеря и т.д.). Гиперосмолярная кома развивается медленно, в течение нескольких дней при беспомощности человека (некомпенсируемая питьем), когда содержание глюкозы достигает 30-50 ммоль/л.
Гипергликемия способствует полиурии, создает гиперосмотическое состояние, которое вызывает дегидратацию тканей, приводящую к нарушению водно-электролитного равновесия.
Резкая дегидротация организма рвотой, диарей, кровопотерей на фоне полиурии и отсутствия питья приводит к гиповолемии. Гиповолемия вызывает снижение АД, сгущение крови, увеличение ее вязкости и способности к тромбообразованию. Нарушение гемодинамики приводит к ишемии тканей, развитию гипоксии, накоплению лактата и энергодефициту. Ишемия почек приводит к развитию острой почечной недостаточности – анурии. Анурия приводит к накоплению в крови остаточного азота (аммиак, мочевина, аминокислоты), возникает гиперазотемия. Гиповолемия через альдостерон снижает выведение с мочой NaCl, что вызывает
гипернатриемию и гиперхлоремию. Гиперазотемия, гипернатриемия и гиперхлоремия усиливают гиперосмотическое состояние и нарушение водно-электролитного равновесия.
Энергодефицит и нарушение водно-электролитного равновесия препятствует формированию на мембране нейронов потенциала и проведению нервных импульсов в ЦНС, что приводит к развитию комы. Смертность при гипергликемической коме 50%.
Лактоацидотическая кома характерна для СД II типа, она возникает при накоплении лактата. В присутствии молочной кислоты резко снижается чувствительность адренорецепторов к катехоламинам, развивается необратимый шок. Появляется метаболическая коагулопатия, проявляющаяся ДВС-синдромом, периферическими тромбозами, тромбоэмболиями (инфаркт миокарда, инсульт).
58
Ацидоз при избытке кетоновых тел и лактата затрудняет отдачу Hb кислорода в ткани (гипоксия), он блокирует активность большинства ферментов, в первую очередь подавляется синтез АТФ, активный транспорт и создание мембранных градиентов, что в нервной ткани угнетает проведение нервных импульсов и вызывает кому.
Поздние осложнения сахарного диабета
Поздние осложнения СД неспецифичны (возникают при разных видах СД), к ним относятся:
1.макроангиопатия (атеросклероз крупных артерий);
2.нефропатия;
3.ретинопатия;
4.нейропатия;
5.синдром диабетической стопы.
Главная причина поздних осложнений сахарного диабета является гипергликемия, гипер-липидемия и гиперхолестеринемия. Они приводят к повреждению кровеносных сосудов и нарушению функций различных органов и тканей путем гликозилирования белков, образова-ния сорбитола и активации атеросклероза.
1. Неферментативное гликозилирование белков. Глюкоза взаимодействует со свободны-ми аминогруппами белков с образованием Шиффовых оснований, при этом белки изменяют свою конформацию и функции. Степень гликозилирования белков зависит от скорости их обновления и концентрации глюкозы.
При гликозилировании кристаллинов - белков хрусталика, образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта.
При гликозилировании белков (протеогликаны, коллагены, гликопротеины) базальных мембран нарушается их обмен, соотношение и структурная организация, происходит утолщение базальных мембран и развитие ангиопатий.
Макроангиопатии проявляются в поражениях крупных и средних сосудов сердца, мозга, нижних конечностей. Гликозилированные белки базальных мембран и межклеточного матрикса (коллагена и эластина) снижают
59
эластичности артерий. Гликозилирование в сочета-нии с гиперлипидемией гликозилированных ЛП и гиперхолестеринемией является причиной активации атеросклероза.
Микроангиопатии — результат повреждения капилляров и мелких сосудов. Проявляются в форме нефро-, нейро- и ретинопатии.
Нефропатия развивается примерно у трети больных СД. Признаком ранних стадий нефропатии служит микроальбуминурия (в пределах 30—300 мг/сут), которая в дальнейшем развивается до классического нефротического синдрома, характеризующегося высокой про-теинурией, гипоальбуминемией и отёками.
Ретинопатия, самое серьёзное осложнение сахарного диабета и наиболее частая причина слепоты, развивается у 60-80% больных СД. На ранних стадиях развивается базальная рети-нопатия, которая проявляется в кровоизлияниях в сетчатку, расширении сосудов сетчатки, отёках. Если изменения не затрагивают жёлтого пятна, потеря зрения обычно не происходит. В дальнейшем может развиться пролиферативная ретинопатия, проявляющаяся в ново-образовании сосудов сетчатки и стекловидного тела. Ломкость и высокая проницаемость но-вообразованных сосудов определяют частые кровоизлияния в сетчатку или стекловидное те-ло. На месте тромбов развивается фиброз, приводящий к отслойке сетчатки и потере зрения.
2. Превращение глюкозы в сорбитол. При гипергликемии этот процесс ускоряется. Реакция катализируется альдозоредуктазой. Сорбитол не используется в клетке, а скорость его диффузии из клеток невелика. При гипергликемии сорбитол накапливается в сетчатке и хру-сталике глаза, клетках клубочков почек, шванновских клетках, в эндотелии. Сорбитол в высоких концентрациях токсичен для клеток, он приводит к увеличению осмотического дав-ления, набуханию клеток и отёку тканей. При накоплении сорбитола в хрусталике приводит к набуханию и нарушению упорядоченной структуры кристаллинов, в результате хрусталик мутнеет.
Диагностика сахарного диабета
Диагноз сахарного диабета ставят на основе классических симптомов сахарного диабета — полиурии, полидипсии, полифагии, ощущения сухости во рту.
Биохимическими признаками СД являются:
•Уровень глюкозы натощак в капиллярной крови выше 6,1 ммоль/л;
60
•Уровень С-пептида натощак менее 0,4 ммоль/л – признак СД I типа.
•Тест с глюкагоном. Натощак определяется концентрация С-пептида (в норме >0,6 ммоль/л), затем 1мг глюкагона вводят внутривенно, через 6 минут определяется концентрация С-пептида (в норме >1,1 ммоль/л).
•Наличие глюкозурии (определяют для контроля лечения);
•Глюкозотелерантный тест (ГТТ), проводится при отсутствии клинических симптомов СД, когда концентрация глюкозы в крови натощак соответствует норме. При-знак СД - уровень глюкозы в плазме крови выше 11,1 ммоль/л через 2 ч после сахарной нагрузки;
Определение толерантности к глюкозе
Обследуемый принимает раствор глюкозы (250-300 мл воды + глюкоза 1 г на 1 кг массы тела). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 — у здорового человека; 2 — у больного сахарным диабетом.
Для оценки компенсации СД определяют:
•В норме уровень гликозилированного гемоглобина НbА1с не более 6% от общего содержания Hb, при компенсированном СД НbА1с < 8,5%;
•альбуминурии. В норме альбуминов в моче < 30 мг/сут. При сахарном диабете до 300 мг/сут.
Поскольку СД II типа развивается значительно медленнее, классические клинические симптомы, гипергликемию и дефицит инсулина диагностируют позднее, часто в сочетании с симптомами поздних осложнений сахарного диабета.
Лечение сахарного диабета
Лечение сахарного диабета зависит от его типа (I или II), является комплексным и включает диету, применение сахаропонижающих средств, инсулинотерапию, а также профилактику и лечение осложнений.
Сахаропонижающие препараты делят на две основные группы: производные сульфонилмочевины и бигуаниды.
61
Препараты сульфонилмочевины блокируют АТФ-чувствительные К+-каналы, что повышает внутриклеточную концентрацию К+ и приводит к деполяризации мембраны. Деполяризация мембраны ускоряет транспорт ионов кальция в клетку, вследствие чего стимулируется секреция инсулина.
Бигуаниды увеличивают количество переносчиков глюкозы ГЛЮТ-4 на поверхности мембран клеток жировой ткани и мышц.
Инсулинотерапия обязательна для СД I типа (1-4 инъекции в день), при СД II типа инсулин иногда назначают для лучшего контроля СД, а также при развитии через 10-15 лет вторичной абсолютной инсулиновой недостаточности.
К перспективным методам лечения сахарного диабета относят следующие: трансплантация островков поджелудочной железы или изолированных β- клеток, трансплантация генетически реконструированных клеток, а также стимуляция регенерации панкреатических островков.
При сахарном диабете обоих типов важнейшее значение имеет диетотерапия. Рекомендуют хорошо сбалансированную диету: на долю углеводов должно приходиться 50—60% общей калорийности пиши (исключение должны составлять легкоусвояемые углеводы, пиво, спиртные напитки, сиропы, пирожные и др.); на долю белков — 15—20%; на долю всех жиров — не более 25-30%. Пищу следует принимать 5—6 раз в течение суток.
31. ИЗСД и ИНЗСД: механизмы развития патохимических нарушений, сходство и отличие в отклонении
биохимических показателей.
САХАРНЫЙ ДИАБЕТ I типа
СД I типа — заболевание, которое возникает вследствие абсолютного дефицита инсули-на, вызванного аутоиммунным разрушением β-клеток поджелудочной железы. СД I типа по-ражает в большинстве случаев детей, подростков и молодых людей до 30 лет, но может про-явиться в любом возрасте. СД I типа редко является семейным заболеванием (10-15% всех случаев).
Причины СД I типа
1. Генетическая предрасположенность. Генетические дефекты ведущие к СД могут реа-лизоваться в клетках иммунной системы и β-клетках поджелудочной железы. В β-клетках известно около 20 генов,
62
способствующих развитию СД I типа. В 60-70% случа-ях СД I типа связан с наличием в 6 хромосоме HLA региона генов DR3, DR4 и DQ.
2. Действие на β-клетки β-цитотропных вирусов (оспа, краснуха, корь, паротит, Кокса-ки, аденовирус, цитомегаловирус), химических и других диабетогенов.
Вариант 1
При наличии генетического дефекта, на поверхности β-клеток накапливаются антиге-ны, имеющие схожую аминокислотную последовательность с β- цитотропными вирусами.
В случае возникновения инфекции β-цитотропных вирусов, развиваются иммунные реакции против этих вирусов и аутоиммунные реакции против схожих антигенов β-клеток. Реакция идет с участием моноцитов, Т- лимфоцитов, антител к β-клеткам, инсулину, глута-мат декарбоксилазе (фермент 64кДа, находиться на мембране β-клеток). В результате аутоиммунные реакции вызывают гибель β-клеток.
Вариант 2
При действии на β-клетки с генотипом HLA β-цитотропных вирусов или диабетогенов на поверхности β-клеток происходит изменение антигенов.
На измененные антигены β-клетки развиваются аутоиммунные реакции. Аутоиммунные реакции вызывают гибель β-клеток.
Вариант 3
β-цитотропные вирусы имеют схожую последовательность аминокислот с глутамат декарбоксилазой β-клеток. Генетический дефект СД8+ лимфоцитов (Т-супрессоров) не позволяет им отличить аминокислотную последовательность вируса и глутамат декарбоксилазы, поэтому при возникновении инфекции, Т-лимфоциты реагируют на глутамат декарбоксилазу β-клеток как на вирус.
Вариант 4
Некоторые β-цитотропные вирусы и химические диабетогены, например, производные нитрозомочевины, нитрозамины, аллоксан самостоятельно и избирательно поражают β-клетки, вызывая их лизис;
Стадии развития СД I типа
63
7.Стадия генетической предрасположенности. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
8.Стадия провоцирующих событий. Инфекция β-цитотропных вирусов или действие химических диабетогенов. Протекает без клинических симптомов;
9.Стадия явных иммунных аномалий. Развитие смешанных аутоиммунных реакций против β-клеток. Ресурсы инсулина достаточны. Протекает без клинических симптомов.
Развивается от 2-3 месяцев до 2-3 лет;
10.Стадия латентного диабета. Гибель 75% β-клеток,
небольшое снижение инсулина, гипергликемия при нагрузочных пробах, снижение аутоиммунных процессов. Протекает без клинических симптомов;
11.Явный диабет. Гибель 80-90% β-клеток, заметное снижение инсулина, гипергликемия натощак, нет или слабые аутоиммунные реакции. Появляются клинические симптомы. Развивается 2 года. Необходима инсулинотерапия;
12.Терминальный диабет. Полная гибель β-клеток, высокая потребность в инсулинотерапии, аутоиммунные проявления снижены или их нет. Выраженные клинические проявления, появляются ангиопатии. Развивается до 3,5 лет;
Изменения метаболизма при СД I типа
При СД I типа исчезает инсулин, т.к. инсулин ингибитор секреции глюкагона, в крови происходит увеличение глюкагона.
Изменения в углеводном обмене
64
Ï Å× ÅÍ Ü |
|
|
|
Ï ÔØ |
|
|
ÊÐÎ ÂÜ |
|
|
|
|
|
|
|
|
||
|
|
ãëè êî ãåí |
Ãëþ êî çà |
|
|
Ãëþ êî çà |
||
Белки |
ÀÊ |
ÀÊ |
Ï ÂÊ |
ÑÆÊ |
|
ÑÆÊ |
||
|
|
|
||||||
|
|
|
NH |
|
|
|
|
|
|
|
|
3 |
Ù ÓÊ |
АцетилКо А |
ÊÒ |
ÊÒ |
|
|
|
|
|
|||||
|
ì |
о чевин а |
ÖÒÊ |
ÕÑ |
|
|
||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
ÄÖ |
|
Í |
ÀÄÍ 2 |
|
ÒÃ |
|
|
|
|
|
|
|
ÕÑ |
|||
|
|
|
|
|
|
|
|
ÒÃ |
|
ÀÒÔ |
|
|
|
|
ËÏ Î |
Í Ï |
ËÏ Î Í Ï |
|
|
|
|
|
|
|
||
|
|
ÀÊ, ì |
î ÷åâè í à |
|
|
|
ÀÊ, ì î ÷åâè í à |
|
|
|
|
|
|
|
|
|
Àçî ò |
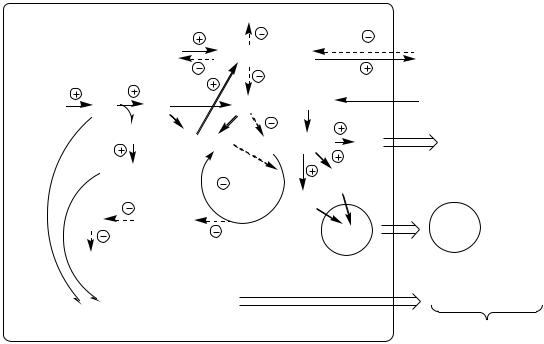
В печени дефицит инсулина и избыток глюкагона стимулирует реакции глюконеогенеза, гликогенолиза и ингибирует реакции гликолиза, ПФШ и синтеза гликогена. В результате в печени глюкозы больше образуется, чем потребляется.
Так как реакции глюконеогенеза протекают через ЩУК, он, образовавшись из ПВК, аспартата и малата, активно вовлекается в глюконеогенез, вместо того чтобы включаться в ЦТК. В результате ЦТК и ДЦ тормозится, снижается образование АТФ, возникает энергодефицит.
В инсулинзависимых тканях (мышцы, жировая ткань) дефицит инсулина препятствует поступлению глюкозы в клетки и ее использованию в реакциях гликолиза, ПФШ и синтеза гликогена. Блокирование ЦТК и ДЦ также вызывает энергодефицит.
Снижение потребления глюкозы инсулинзависимыми тканями и усиление ее образования в печени приводит к гипергликемии. Когда гипергликемия превышает концентрационный почечный порог возникает глюкозурия.
Глюкозурия – наличие глюкозы моче. В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу. Если уровень глюкозы превышает в крови 9-10 ммоль/л, глюкоза не успевает полностью реабсорбироваться из первичной мочи и частично выводится с вторичной мочой.
65
У больных с СД после приёма пищи концентрация глюкозы в крови может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе.
ÀÄÈ Ï Î ÖÈ Ò |
|
|
ÊÐÎ ÂÜ |
|
|
|
|
|
|
|
Ãëþ êî çà |
|
Ãëþ êî çà |
|
ÒÃ |
ÆÊ |
|
ÆÊ |
|
|
глицеро -3Ф |
|
ãë è öåðî -3Ô |
|
|
|
|
|
|
|
|
|
|
Деф ицит ин сулин а |
|
|
ÆÊ |
ËÏ Ë |
ÕÑ |
|
|
|
||
|
|
|
ÒÃ |
|
|
|
|
|
|
|
|
|
|
ËÏ Î Í Ï |
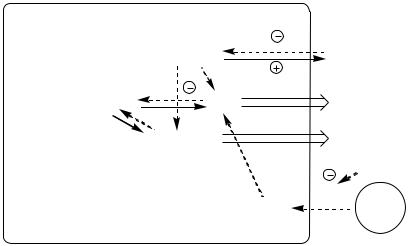
Изменения в липидном обмене
Дефицит АТФ, НАДФН2, инсулина и избыток глюкагона тормозят липогенез и усиливают липолиз в жировой ткани. В результате в крови повышается концентрация свободных жирных кислот, которые поступают в печень и окисляются там до Ацетил-КоА. АцетилКоА при дефиците ЩУК не может включаться в ЦТК. Поэтому он накапливается и поступает на альтернативные пути: синтез кетоновых тел (ацетоуксусная, β-гидроксимасляная кислоты) и холестерина.
Внорме кетоновые тела являются источником энергии для аэробных тканей, они превращаются в АцетилКоА, который окисляется в ЦТК. Так как ЦТК заблокирован дефицитом ЩУК, кетоновые тела накапливаются в крови и вызывают кетонемию. Кетонемия усугубляет недостаточность инсулина, подавляя остаточную секреторную активность β-клеток. Когда кетонемия превышает концентрационный почечный порог (выше 20 мг/дл, иногда до 100 мг/дл) возникает кетонурия. Кетонурия – наличие кетоновых тел в моче.
Втканях ацетоуксусная кислота частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии.
Липопротеины крови поставляют субстраты для липогенеза в тканях. Дефицит инсулина блокирует липогенез в жировой ткани, ингибирует липопротеинлипазу в сосудах, это препятствует расщеплению липопротеинов крови (в основном, ЛПОНП), в результате они накапливаются,
вызывая гиперлипопротеинемию.
66
Изменения в белковом обмене
Энергодефицит, недостаток инсулина и избыток глюкагона приводит к снижению скорости синтеза белков в организме и усилению их распада, что повышает концентрацию аминокислот в крови. Аминокислоты поступают в печень и дезаминируются до кетокислот. Кетокислоты включаются в глюконеогенез, что усиливает гипергликемию. Из аммиака активно синтезируется мочевина. Повышение в крови аммиака, мочевины, аминокислот вызывает азотемию – увеличение концентрации азота в крови. Азотемия приводит к азотурии – увеличению концентрации азота в моче. Развивается отрицательный азотистый баланс. Катаболизм белков ведет к миодистрофии и вторичному иммунодефициту.
Изменения в водно-солевом обмене
Поскольку возможности почек ограничены, высокие концентрации глюкозы, кетоновых тел и мочевины не успевают реабсорбироваться из первичной мочи. Они создают в первичной моче высокое осмотическое давление, которое препятствует реабсорбции воды в кровь и образованию вторичной мочи. У таких пациентов развивается полиурия, выделение мочи возрастает до 3—4 л в сутки (в некоторых случаях до 8—9 л). Потеря воды вызывает постоянную жажду или полидипсию. Без частого питья, полиурия может приводить к обезвоживанию организма. Потеря с мочой глюкозы усугубляет энергодефицит, может увеличить аппетит и полифагию. С первичной мочой из организма уходят некоторые полезные минеральные компоненты, что приводит к нарушению минерального обмена.
Высокие концентрации глюкозы, кетоновых тел и мочевины создают в плазме крови значительное осмотическое давление, которое способствует дегидратации тканей. Кроме воды ткани теряют электролиты, прежде всего ионы К+, Na+, С1-, НСО3-.
Изменение в газообмене тканей
Общая дегидратация организма, вызванная полиурией и дегидратацией тканей приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Причиной гипоксии является также гликозилирование Hb в HbA1c, который не переносит О2 к тканям. Гипоксия ведет к энергодефициту и накоплению в организме
лактата.
Изменения в кислотно-основном равновесии
67

Накопление кетоновых тел, лактата и потеря щелочных валентностей с мочой снижает буферную ёмкость крови и вызывает ацидоз.
Симптомы СД I типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены. Общая слабость, похудание, снижение трудоспособности, сонливость. Ожирение отсутствует. Повышенный аппетит при кетоацидозе сменяется анорексией. Развивается быстро, склонен к развитию кетоацидотической комы.
САХАРНЫЙ ДИАБЕТ II типа
СД II типа представляет собой группу гетерогенных нарушений углеводного обмена. СД II типа не инсулинозависимый, не склонен к кетоацидотической коме, не имеет антител к β-клеткам, не аутоиммунной природы, не имеет связи с определенными HLA фенотипами. Ожирение в 80%. На долю СД II типа приходится примерно 85-90% всех случаев СД, он поражает людей, как правило, старше 40 лет и характеризуется высокой частотой семейных форм (риск СД II типа у ближайших родственников больного достигает 50%, тогда как при СД I типа он не превышает 10%). СД II типа поражает преимущественно жителей развитых стран, особенно горожан.
Воснове СД II типа лежат множество причин. СД II типа развивается при:
генетических дефектах рецепторов инсулина, у них снижается чувствительность к инсулину;
синтезе дефектного инсулина с низкой биологической активностью (мутация гена инсулина: в позиции 24 В-цепи вместо фен присутствует лей);
нарушении превращения проинсулина в инсулин;
нарушении секреции инсулина;
повреждении инсулина и его рецепторов антителами;
повышения скорости катаболизма инсулина;
действия контринсулярных гормонов (создают гипеинсулинемию, которая вызывает инсулинорезистентность);
нарушении глюкозочувствительного механизма  клеток (мутации гена глюкокиназы) и т.д.
клеток (мутации гена глюкокиназы) и т.д.
Основным провоцирующим фактором СД II типа служит ожирение.
68
Стадии СД II типа
4.Стадия генетической предрасположенности. Есть генетические маркеры, нет нарушений углеводного обмена. Может длиться всю жизнь;
5.Стадия латентного диабета. Гипергликемия при нагрузочных пробах. Протекает без клинических симптомов СД;
6.Явный диабет. Гипергликемия натощак. Появляются клинические симптомы.
Симптомы СД II типа
Общие симптомы (жажда, полиурия, кожный зуд, склонность к инфекциям) выражены умеренно или отсутствуют. Часто ожирение (у 80-90% больных).
Изменения метаболизма при СД II типа
Относительный дефицит инсулина вызывает метаболические нарушения, схожие с теми которые возникают при абсолютном дефиците инсулина, однако эти нарушения менее выражены, а у 50% больных с ожирением и умеренной гипергликемией СД II типа вообще протекает бессимптомно.
В отличие от абсолютного дефицита инсулина, при относительном дефиците инсулина, влияние инсулина сохраняется на жировую ткань, имеющую высокое содержание рецепторов к инсулину. Инсулин в жировой ткани стимулирует липогенез, блокирует липолиз и выход жирных кислот в кровь, поэтому при СД II типа не наблюдается кетоацидоз, масса тела не уменьшается, а наоборот развивается ожирение. Таким образом, ожирение, с одной стороны, важнейший фактор риска, а с другой — одно из ранних проявлений СД II типа.
При СД II типа наблюдается гиперинсулинемия (80%), артериальная гипертензия (50%), гиперлипидемия (50%), атеросклероз, нейропатия (15%) и диабетическая нефропатия (5%).
Осложнения СД
Острые осложнения сахарного диабета. Механизмы развития диабетической комы
69
Острые осложнения специфичны для СД I и II типа.
Дегидратация тканей головного мозга в первую очередь, а также нарушения обмена веществ в нервной ткани могут приводить к развитию острых осложнений в виде коматозных состояний. Кома это крайне тяжелое состояние, характеризующееся глубоким угнетением ЦНС, стойкой потерей сознания, утратой реакций на внешние раздражители любой интенсивности. Коматозные состояния при СД могут проявляться в трёх формах: кетоацидотической, гиперосмолярной и лактоацидотической.
Кетоацидотическая кома возникает при СД I типа, когда концентрация кетоновых тел становится выше 100 мг/дл (до 400-500мг/дл).
Гиперкетонемия приводит к:
1)ацидозу, который блокирует активность большинства ферментов, в первую дыхательных, что вызывает гипоксию и снижение синтеза АТФ.
2)гиперосмолярности, которая приводит к дегидратации тканей и нарушению водно-электролитного равновесия, с потерей ионов калия, натрия, фосфора, магния, кальция, бикарбонатов.
Это при определенной выраженности и вызывает коматозное состояние с падением артериального давления и развитием острой почечной недостаточности.
Возникающая гипокалиемия ведет к гипотонии гладкой и поперечнополосатой мускулатуры, снижению тонуса сосудов, падению АД, сердечной аритмии, гипотонии дыхательной мускулатуры с развитием острой дыхательной недостаточности; атонии ЖКТ с парезом желудка и развитием кишечной непроходимости развивается выраженная гипоксия. В общей причине смертности она занимает 2-4 %.
Гиперосмолярная кома характерна для СД II типа, она наблюдается при высокой гипергликемии. У большинства высокая гипергликемия обусловлена сопутствующим нарушением функции почек, ее провоцируют стресс, травма, резкая дегидратация организма (рвота, диарея, ожоги, кровопотеря и т.д.). Гиперосмолярная кома развивается медленно, в течение нескольких дней при беспомощности человека (некомпенсируемая питьем), когда содержание глюкозы достигает 30-50 ммоль/л.
70
Гипергликемия способствует полиурии, создает гиперосмотическое состояние, которое вызывает дегидратацию тканей, приводящую к нарушению водно-электролитного равновесия.
Резкая дегидротация организма рвотой, диарей, кровопотерей на фоне полиурии и отсутствия питья приводит к гиповолемии. Гиповолемия вызывает снижение АД, сгущение крови, увеличение ее вязкости и способности к тромбообразованию. Нарушение гемодинамики приводит к ишемии тканей, развитию гипоксии, накоплению лактата и энергодефициту. Ишемия почек приводит к развитию острой почечной недостаточности – анурии. Анурия приводит к накоплению в крови остаточного азота (аммиак, мочевина, аминокислоты), возникает гиперазотемия. Гиповолемия через альдостерон снижает выведение с мочой NaCl, что вызывает
гипернатриемию и гиперхлоремию. Гиперазотемия, гипернатриемия и гиперхлоремия усиливают гиперосмотическое состояние и нарушение водно-электролитного равновесия.
Энергодефицит и нарушение водно-электролитного равновесия препятствует формированию на мембране нейронов потенциала и проведению нервных импульсов в ЦНС, что приводит к развитию комы. Смертность при гипергликемической коме 50%.
Лактоацидотическая кома характерна для СД II типа, она возникает при накоплении лактата. В присутствии молочной кислоты резко снижается чувствительность адренорецепторов к катехоламинам, развивается необратимый шок. Появляется метаболическая коагулопатия, проявляющаяся ДВС-синдромом, периферическими тромбозами, тромбоэмболиями (инфаркт миокарда, инсульт).
Ацидоз при избытке кетоновых тел и лактата затрудняет отдачу Hb кислорода в ткани (гипоксия), он блокирует активность большинства ферментов, в первую очередь подавляется синтез АТФ, активный транспорт и создание мембранных градиентов, что в нервной ткани угнетает проведение нервных импульсов и вызывает кому.
Поздние осложнения сахарного диабета
Поздние осложнения СД неспецифичны (возникают при разных видах СД), к ним относятся:
6.макроангиопатия (атеросклероз крупных артерий);
7.нефропатия;
71
8.ретинопатия;
9.нейропатия;
10.синдром диабетической стопы.
Главная причина поздних осложнений сахарного диабета является гипергликемия, гипер-липидемия и гиперхолестеринемия. Они приводят к повреждению кровеносных сосудов и нарушению функций различных органов и тканей путем гликозилирования белков, образова-ния сорбитола и активации атеросклероза.
1. Неферментативное гликозилирование белков. Глюкоза взаимодействует со свободны-ми аминогруппами белков с образованием Шиффовых оснований, при этом белки изменяют свою конформацию и функции. Степень гликозилирования белков зависит от скорости их обновления и концентрации глюкозы.
При гликозилировании кристаллинов - белков хрусталика, образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта.
При гликозилировании белков (протеогликаны, коллагены, гликопротеины) базальных мембран нарушается их обмен, соотношение и структурная организация, происходит утолщение базальных мембран и развитие ангиопатий.
Макроангиопатии проявляются в поражениях крупных и средних сосудов сердца, мозга, нижних конечностей. Гликозилированные белки базальных мембран и межклеточного матрикса (коллагена и эластина) снижают эластичности артерий. Гликозилирование в сочета-нии с гиперлипидемией гликозилированных ЛП и гиперхолестеринемией является причиной активации атеросклероза.
Микроангиопатии — результат повреждения капилляров и мелких сосудов. Проявляются в форме нефро-, нейро- и ретинопатии.
Нефропатия развивается примерно у трети больных СД. Признаком ранних стадий нефропатии служит микроальбуминурия (в пределах 30—300 мг/сут), которая в дальнейшем развивается до классического нефротического синдрома, характеризующегося высокой про-теинурией, гипоальбуминемией и отёками.
72
Ретинопатия, самое серьёзное осложнение сахарного диабета и наиболее частая причина слепоты, развивается у 60-80% больных СД. На ранних стадиях развивается базальная рети-нопатия, которая проявляется в кровоизлияниях в сетчатку, расширении сосудов сетчатки, отёках. Если изменения не затрагивают жёлтого пятна, потеря зрения обычно не происходит. В дальнейшем может развиться пролиферативная ретинопатия, проявляющаяся в ново-образовании сосудов сетчатки и стекловидного тела. Ломкость и высокая проницаемость но-вообразованных сосудов определяют частые кровоизлияния в сетчатку или стекловидное те-ло. На месте тромбов развивается фиброз, приводящий к отслойке сетчатки и потере зрения.
2. Превращение глюкозы в сорбитол. При гипергликемии этот процесс ускоряется. Реакция катализируется альдозоредуктазой. Сорбитол не используется в клетке, а скорость его диффузии из клеток невелика. При гипергликемии сорбитол накапливается в сетчатке и хру-сталике глаза, клетках клубочков почек, шванновских клетках, в эндотелии. Сорбитол в высоких концентрациях токсичен для клеток, он приводит к увеличению осмотического дав-ления, набуханию клеток и отёку тканей. При накоплении сорбитола в хрусталике приводит к набуханию и нарушению упорядоченной структуры кристаллинов, в результате хрусталик мутнеет.
Диагностика сахарного диабета
Диагноз сахарного диабета ставят на основе классических симптомов сахарного диабета — полиурии, полидипсии, полифагии, ощущения сухости во рту.
Биохимическими признаками СД являются:
•Уровень глюкозы натощак в капиллярной крови выше 6,1 ммоль/л;
•Уровень С-пептида натощак менее 0,4 ммоль/л – признак СД I типа.
•Тест с глюкагоном. Натощак определяется концентрация С-пептида (в норме >0,6 ммоль/л), затем 1мг глюкагона вводят внутривенно, через 6 минут определяется концентрация С-пептида (в норме >1,1 ммоль/л).
•Наличие глюкозурии (определяют для контроля лечения);
•Глюкозотелерантный тест (ГТТ), проводится при отсутствии клинических симптомов СД, когда концентрация глюкозы в крови натощак соответствует норме. При-знак СД - уровень глюкозы в плазме крови выше 11,1 ммоль/л через 2 ч после сахарной нагрузки;
73

Определение толерантности к глюкозе
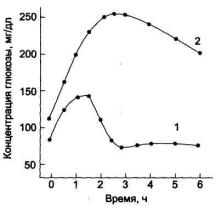
Обследуемый принимает раствор глюкозы (250-300 мл воды + глюкоза 1 г на 1 кг массы тела). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 — у здорового человека; 2 — у больного сахарным диабетом.
Для оценки компенсации СД определяют:
•В норме уровень гликозилированного гемоглобина НbА1с не более 6% от общего содержания Hb, при компенсированном СД НbА1с < 8,5%;
•альбуминурии. В норме альбуминов в моче < 30 мг/сут. При сахарном диабете до 300 мг/сут.
Поскольку СД II типа развивается значительно медленнее, классические клинические симптомы, гипергликемию и дефицит инсулина диагностируют позднее, часто в сочетании с симптомами поздних осложнений сахарного диабета.
Лечение сахарного диабета
Лечение сахарного диабета зависит от его типа (I или II), является комплексным и включает диету, применение сахаропонижающих средств, инсулинотерапию, а также профилактику и лечение осложнений.
Сахаропонижающие препараты делят на две основные группы: производные сульфонилмочевины и бигуаниды.
Препараты сульфонилмочевины блокируют АТФ-чувствительные К+-каналы, что повышает внутриклеточную концентрацию К+ и приводит к деполяризации мембраны. Деполяризация мембраны ускоряет транспорт ионов кальция в клетку, вследствие чего стимулируется секреция инсулина.
Бигуаниды увеличивают количество переносчиков глюкозы ГЛЮТ-4 на поверхности мембран клеток жировой ткани и мышц.
Инсулинотерапия обязательна для СД I типа (1-4 инъекции в день), при СД II типа инсулин иногда назначают для лучшего контроля СД, а также при развитии через 10-15 лет вторичной абсолютной инсулиновой недостаточности.
74
К перспективным методам лечения сахарного диабета относят следующие: трансплантация островков поджелудочной железы или изолированных β- клеток, трансплантация генетически реконструированных клеток, а также стимуляция регенерации панкреатических островков.
При сахарном диабете обоих типов важнейшее значение имеет диетотерапия. Рекомендуют хорошо сбалансированную диету: на долю углеводов должно приходиться 50—60% общей калорийности пиши (исключение должны составлять легкоусвояемые углеводы, пиво, спиртные напитки, сиропы, пирожные и др.); на долю белков — 15—20%; на долю всех жиров — не более 25-30%. Пищу следует принимать 5—6 раз в течение суток.
32. Глюкозотолерантный тест, методика проведения, диагностическое значение. Биохимическая лабораторная
диагностика состояния углеводного обмена
Глюкозотелерантный тест (ГТТ), проводится при отсутствии клинических симптомов СД, когда концентрация глюкозы в крови натощак соответствует норме. Признак СД - уровень глюкозы в плазме крови выше 11,1 ммоль/л через 2 ч после сахарной нагрузки;
Определение толерантности к глюкозе
Обследуемый принимает раствор глюкозы (250-300 мл воды + глюкоза 1 г на 1 кг массы тела). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 — у здорового человека; 2 — у больного сахарным диабетом.
33.Важнейшие липиды пищи и организма человека: классификация, физико-химические свойства,
биологическая роль. Принципы нормирования суточной потребности липидов в разные периоды детства и взрослого.
КЛАССИФИКАЦИЯ ЛИПИДОВ
Липиды по способности к гидролизу делят на омыляемые (двух и более компонентные) и неомыляемые (однокомпонентные).
Омыляемые липиды в щелочной среде гидролизуются с образованием мыл, они содержат в своем в составе жирные кислоты и спирты глицерин
75
(глицеролипиды) или сфингозин (сфинголипиды). По количеству компонентов омыляемые липиды делятся на простые (состоят из 2 классов соединений) и сложные (состоят из 3 и более классов).
К простым липидам относятся:
1)воска (сложный эфир высшего одноатомного спирта и жирной кислоты);
2)триацилглицериды, диацилглицериды, моноацилглицериды (сложный эфир глицерина и жирных кислот). У человека весом в 70 кг ТГ около 10 кг.
3)церамиды (сложный эфир сфингозина и жирной кислоты С18-26) – лежат в основе сфинголипидов;
К сложным липидам относятся:
1) фосфолипиды (содержат фосфорную кислоту):
а) фосфоглицеролипиды (сложный эфир глицерина и 2 жирных кислот, содержит фосфорную кислоту и аминоспирт) - фосфатидилсерин, фосфатидилэтаноламин, фосфатидилхолин, фосфатидилинозитол, фосфатидилглицерол;
б) кардиолипины (2 фосфатидные кислоты, соединенные через глицерин);
в) плазмалогены (сложный эфир глицерина и жирной кислоты, содержит ненасыщенный одноатомный высший спирт, фосфорную кислоту и аминоспирт) – фосфатидальэтаноламины, фосфатидальсерины, фосфатидальхолины;
г) сфингомиелины (сложный эфир сфингозина и жирной кислоты С18-26, содержит фосфорную кислоту и аминоспирт - холин);
2) гликолипиды (производные сфингозина, содержащие углеводы):
а) цереброзиды (сложный эфир сфингозина и жирной кислоты С18-26, содержит гексозу: глюкозу или галактозу);
б) сульфатиды (сложный эфир сфингозина и жирной кислоты С18-26, содержит гексозу (глюкозу или галактозу) к которой присоединена в 3 положение серная кислота). Много в белом веществе;
в) ганглиозиды (сложный эфир сфингозина и жирной кислоты С18-26, содержит олигосахарид из гексоз и сиаловых кислот). Находятся в ганглиозных клетках;
К неомыляемым липидам относят:
76
1.стероиды;
2.жирные кислоты (структурный компонент омыляемых липидов),
3.витамины А, Д, Е, К;
4.терпены (углеводороды, спирты, альдегиды и кетоны с несколькими звеньями изопрена).
БИОЛОГИЧЕСКИЕ ФУНКЦИИ ЛИПИДОВ
Ворганизме липиды выполняют разнообразные функции:
1)Структурная. Сложные липиды и холестерин амфифильны, они образуют все клеточные мембраны; фосфолипиды выстилают поверхность альвеол, образуют оболочку липопротеинов.
Сфингомиелины, плазмалогены, гликолипиды образуют миелиновые оболочки и другие мембраны нервных тканей.
2)Энергетическая. В организме до 33% всей энергии АТФ образуется за счет окисления липидов;
3)Антиоксидантная. Витамины А, Д, Е, К препятсвуют СРО;
4)Запасающая. ТГ являются формой хранения жирных кислот;
5)Защитная. ТГ, в составе жировой ткани, обеспечивают теплоизоляционную и механическую защиту тканей. Воска образуют защитную смазку на коже человека;
6)Регуляторная. Фосфотидилинозитолы являются внутриклеточными посредниками в действии гормонов (инозитолтрифосфатная система). Из полиненасыщенных жирных кислот образуются эйкозаноиды (лейкотриены, тромбоксаны, простагландины, простациклины), вещества, регулирующие иммуногенез, гемостаз, неспецифическую резистентность организма, воспалительные, аллергические, пролиферативные реакции. Из холестерина образуются стероидные гормоны: половые, кортикоиды, кальцитриол;
7)Пищеварительная. Из холестерина синтезируются желчные кислоты. Желчные кислоты, фосфолипиды, холестерин обеспечивают эмульгирование и всасывание липидов;
8)Информационная. Ганглиозиды обеспечивают межклеточные контакты.
77
Источником липидов в организме являются синтетические процессы и пища. Некоторые липиды в организме не синтезируются (полиненасыщенные жирные кислоты - витамин F, витамины А, Д, Е, К), они являются незаменимыми и поступают в организм только с пищей.
ПРИНЦИПЫ НОРМИРОВАНИЯ ЛИПИДОВ В ПИТАНИИ
В сутки человеку требуется потреблять 80-100г липидов, из них 25-30г растительного масла, 30-50г сливочного масла и 20-30г жира животного происхождения.
Потребность в пищевых липидах зависит от возраста. Новорожденным до 3 месяцев требуется 6,5 г/кг липидов, детям до 6 месяцев - 6 г/кг, детям после 6 месяцев – 5,5 г/кг, взрослым – 1,4 г/кг, пожилым – 0,5 г/кг. Причины: 1).
основным источником энергии для детей грудного возраста являются липиды, а для взрослых людей - глюкоза. 2). Энергозатраты с возрастом снижаются.
Потребность в липидах увеличивается на холоде, при физических нагрузках, в период выздоровления и при беременности.
С пищей в норме поступает около 85-90г ТГ, 1г ФЛ, 0,3—0,5 г ХС (в основном в виде эфиров). Растительные масла содержат много полиеновых незаменимых (линолевая до 60%, линоленовая) жирных кислот, фосфолипидов (удаляются при рафинировании). Сливочное масло содержит много витаминов А, Д, Е.
Все природные липиды хорошо перевариваются, масла усваиваются лучше жиров. При смешанном питании сливочное масло усваивается на 93-98%, свиной жир - на 96-98%, говяжий жир – на 80-94%, подсолнечное масло – на 86-90%. Длительная тепловая обработка (> 30 мин) разрушает полезные липиды, при этом образуются токсические продукты окисления жирных кислот и канцерогенные вещества.
При недостаточном поступлении липидов с пищей снижается иммунитет, снижается продукция стероидных гормонов, нарушается половая функция. При дефиците линолевой кислоты развивается тромбоз сосудов и увеличивается риск раковых заболеваний. При избытке липидов в пище развивается атеросклероз и увеличивается риск рака молочной железы и толстой кишки.
78
34.Переваривание липидов в желудочно-кишечном тракте: роль гормонов, ферментов, желчных кислот. Понятие:
энтерогепатическая циркуляция
ПЕРЕВАРИВАНИЕ ЛИПИДОВ
Переваривание – это гидролиз пищевых веществ до их ассимилируемых форм.
Лишь 40-50% пищевых липидов расщепляется полностью, от 3% до 10% пищевых липи-дов всасываются в неизмененном виде.
Так как липиды не растворимы в воде, их переваривание и всасывание имеет свои осо-бенности и протекает в несколько стадий:
1)Липиды твердой пищи при механическом воздействии и под влиянием ПАВ желчи сме-шиваются с пищеварительными соками с образованием эмульсии (масло в воде). Образо-вание эмульсии необходимо для увеличения площади действия ферментов, т.к. они рабо-тают только в водной фазе. Липиды жидкой пищи (молоко, бульон и т.д.) поступают в организм сразу в виде эмульсии;
2)Под действием липаз пищеварительных соков происходит гидролиз липидов эмульсии с образованием водорастворимых веществ и более простых липидов;
3)Выделенные из эмульсии водорастворимые вещества всасываются и поступают в кровь. Выделенные из эмульсии более простые липиды, соединяясь с компонентами желчи, обра-зуют мицеллы;
4)Мицеллы обеспечивают всасывание липидов в клетки эндотелия кишечника.
Ротовая полость
В ротовой полости происходит механическое измельчение твердой пищи и смачивание ее слюной (рН=6,8).
У грудных детей здесь начинается гидролиз ТГ с короткими и средними жирными кисло-тами, которые поступают с жидкой пищей в виде эмульсии. Гидролиз осуществляет линг-вальная триглицеридлипаза («липаза языка», ТГЛ), которую секретируют железы Эбнера, находящиеся на дорсальной поверхности языка.
79
Желудок
Так как «липаза языка» действует в диапазоне 2-7,5 рН, она может функционировать в же-лудке в течение 1-2 часов, расщепляя до 30% триглицеридов с короткими жирными кислота-ми. У грудных детей и детей младшего возраста она активно гидролизует ТГ молока, которые содержат в основном жирные кислоты с короткой и средней длиной цепей (4—12 С). У взрослых людей вклад «липазы языка» в переваривание ТГ незначителен.
В главных клетках желудка вырабатывается желудочная липаза, которая активна при нейтральном значении рН, характерном для желудочного сока детей грудного и младшего возраста, и не активна у взрослых (рН желудочного сока ~1,5). Эта липаза гидролизует ТГ, отщепляя, в основном, жирные кислоты у третьего атома углерода глицерола. Образующиеся в желудке ЖК и МГ далее участвуют в эмульгировании липидов в двенадцатиперстной киш-ке.
Тонкая кишка
Основной процесс переваривания липидов происходит в тонкой кишке.
1. Эмульгирование липидов (смешивание липидов с водой) происходит в тонкой кишке под действием желчи. Желчь синтезируется в печени, концентрируется в желчном пузыре и после приёма жирной пищи выделяется в просвет двенадцатиперстной кишки (500-1500 мл/сут).
Жёлчь это вязкая жёлто-зелёная жидкость, имеет рН=7,3-8.0, содержит Н2О – 87-97%, ор-ганические вещества (желчные кислоты – 310 ммоль/л (10,3-91,4 г/л), жирные кислоты – 1,4-3,2 г/л, пигменты желчные – 3,2 ммоль/л (5,3-9,8 г/л), холестерин – 25 ммоль/л (0,6-2,6) г/л, фосфолипиды – 8 ммоль/л) и минеральные компоненты (натрий 130-145 ммоль/л, хлор 75-100 ммоль/л, НСО3- 10-28 ммоль/л, калий 5-9 ммоль/л). Нарушение соотношение компонентов желчи приводит к образованию камней.
Жёлчные кислоты (производные холановой кислоты) синтезируются в печени из холе-стерина (холиевая, и хенодезоксихолиевая кислоты) и образуются в кишечнике (дезоксихоли-евая, литохолиевая, и д.р. около 20) из холиевой и хенодезоксихолиевой кислот под действи-ем микроорганизмов.
80
В желчи желчные кислоты присутствуют в основном в виде конъюгатов с глицином (66-80%) и таурином (20-34%), образуя парные желчные кислоты: таурохолевую, гликохолевую и д.р.
Соли жёлчных кислот, мыла, фосфолипиды, белки и щелочная среда желчи действуют как детергенты (ПАВ), они снижают поверхностное натяжение липидных капель, в результате крупные капли распадаются на множество мелких, т.е. происходит эмульгирование. Эмуль-гированию также способствует перистальтика кишечника и выделяющийся, при взаимодействии химуса и бикарбонатов СО2: Н+ + НСО3- → Н2СО3 → Н2О + ↑СО2.
2. Гидролиз триглицеридов осуществляет панкреатическая липаза. Ее оптимум рН=8, она гидролизует ТГ преимущественно в положениях 1 и 3, с образованием 2 свободных жирных кислот и 2-моноацилглицерола (2-МГ). 2- МГ является хорошим эмульгатором.
28% 2-МГ под действием изомеразы превращается в 1-МГ. Большая часть 1- МГ гидролизу-ется панкреатической липазой до глицерина и жирной кислоты.
В поджелудочной железе панкреатическая липаза синтезируется вместе с белком колипа-зой. Колипаза образуется в неактивном виде и в кишечнике активируется трипсином путем частичного протеолиза. Колипаза своим гидрофобным доменом связывается с поверхностью липидной капли, а гидрофильным способствует максимальному приближению активного центра панкреатической липазы к ТГ, что ускоряет их гидролиз.
3. Гидролиз лецитина происходит с участием фосфолипаз (ФЛ): А1, А2, С, D и лизофос-фолипазы (лизоФЛ).
В результате действия этих четырех ферментов фосфолипиды расщепляются до свободных жирных кислот, глицерола, фосфорной кислоты и аминоспирта или его аналога, например, аминокислоты серина, однако часть фосфолипидов расщепляется при участии фосфолипазы А2 только до лизофосфолипидов и в таком виде может поступать в стенку кишечника.
ФЛ А2 активируется частичным протеолизом с участием трипсина и гидролизует лецитин до лизолецитина. Лизолецитин является хорошим
81
эмульгатором. ЛизоФЛ гидролизует часть лизолецитина до глицерофосфохолина. Остальные фосфолипиды не гидролизуются.
4.Гидролиз эфиров холестерина до холестерина и жирных кислот осуществляет холесте-ролэстераза, фермент поджелудочной железы и кишечного сока.
5.Мицеллообразование
Водонерастворимые продукты гидролиза (жирные кислоты с длинной цепью, 2-МГ, холе-стерол, лизолецитины, фосфолипиды) вместе с компонентами желчи (солями жёлчных кис-лот, ХС, ФЛ) образуют в просвете кишечника структуры, называемые смешанными мицелла-ми. Смешанные мицеллы построены таким образом, что гидрофобные части молекул обраще-ны внутрь мицеллы (жирные кислоты, 2-МГ, 1-МГ), а гидрофильные (желчные кислоты, фос-фолипиды, ХС) — наружу, поэтому мицеллы хорошо растворяются в водной фазе содержи-мого тонкой кишки. Стабильность мицелл обеспечивается в основном солями жёлчных кис-лот, а также моноглицеридами и лизофосфолипидами.
Регуляция переваривания
Пища стимулирует секрецию из клеток слизистой тонкой кишки в кровь холецистокини-на (панкреозимин, пептидный гормон). Он вызывает выделение в просвет двенадцатиперст-ной кишки желчи из желчного пузыря и панкреатического сока из поджелудочной железы.
Кислый химус стимулирует секрецию из клеток слизистой тонкой кишки в кровь секре-тина (пептидный гормон). Секретин стимулирует секрецию бикарбоната (НСО3-) в сок под-желудочной железы.
Особенность переваривания липидов у детей
Секреторный аппарат кишечника к моменту рождения ребенка в целом сформирован, в кишечном соке находятся те же ферменты, что и у взрослых, но активность их низкая. Осо-бенно напряженно идет процесс переваривания жиров из-за низкой активности липолитиче-ских ферментов. У детей, находящихся на грудном вскармливании, эмульгированные желчью липиды на 50% расщепляются под влиянием липазы материнского молока.
82
Переваривание липидов жидкой пищи
ВСАСЫВАНИЕ ПРОДУКТОВ ГИДРОЛИЗА
1. Водорастворимые продукты гидролиза липидов всасываются в тонкой кишке без уча-стия мицелл. Холин и этаноламин всасываются в виде ЦДФ производных, фосфорная кислота - в виде Na+ и K+ солей, глицерол - в свободном виде.
2. Жирные кислоты с короткой и средней цепью, всасываются без участия мицелл в основном в тонкой кишке, а часть уже в желудке.
3. Водонерастворимые продукты гидролиза липидов всасываются в тонкой кишке с уча-стием мицелл. Мицеллы сближаются со щёточной каймой энтероцитов, и липидные компо-ненты мицелл (2-МГ, 1-МГ, жирные кислоты, холестерин, лизолецитин, фосфолипиды и т.д.) диффундируют через мембраны внутрь клеток.
Рециклирование компоненты желчи
Вместе с продуктами гидролиза всасываются компоненты желчи - соли жёлчных кислот, фосфолипиды, холестерин. Наиболее активно соли жёлчных кислот всасываются в под-вздошной кишке. Жёлчные кислоты далее попадают через воротную вену в печень, из печени вновь секретируются в жёлчный пузырь и далее опять участвуют в эмульгировании липидов. Этот путь жёлчных кислот называют «энтерогепатическая циркуляция». Каждая молекула жёлчных кислот за сутки проходит 5— 8 циклов, и около 5% жёлчных кислот выделяется с фекалиями.
НАРУШЕНИЯ ПЕРЕВАРИВАНИЯ И ВСАСЫВАНИЯ ЛИПИДОВ. СТЕАТОРЕЯ
Нарушение переваривания липидов может быть при:
1) нарушение оттока жёлчи из жёлчного пузыря (желчекаменная болезнь, опухоль). Уменьшение секреции жёлчи вызывает нарушение эмульгирования липидов, что ведет к снижению гидролиза липидов пищеварительными ферментами;
2) нарушение секреции сока поджелудочной железы приводит к дефициту панкреатиче-ской липазы и снижает гидролиз липидов.
Нарушение переваривания липидов тормозит их всасывание, что приводит к увеличению количества липидов в фекалиях — возникает стеаторея (жирный стул). В норме в фекалиях липидов не более 5%. При стеаторее нарушается
83
всасывание жирорастворимых витаминов (A, D, Е, К) и незаменимых жирных кислот (витамин F), поэтому развиваются гиповитамино-зы жирорастворимых витаминов. Избыток липидов связывает вещества нелипидной природы (белки, углеводы, водорастворимые витамины), и препятствует их перевариванию и всасыва-нию. Возникают гиповитаминозы по водорастворимым витаминам, белковое и углеводное голодание. Непереваренные белки подвергаются гниению в толстой кишке.
35. Транспортные липопротеиды крови классификация (по плотности, электрофоретической подвижности, по апопротеинам), место синтеза, функции, диагностическое значение (а – г): хиломикроны (ХМ), обмен хиломикронов в абсорбтивный период липопротеины очень низкой (ЛПОНП) обмен в постабсорбтивный период
липопротеины низкой плотности (ЛПНП) липопротеины высокой плотности (ЛПВП)
ТРАНСПОРТ ЛИПИДОВ В ОРГАНИЗМЕ
Транспорт липидов в организме идет двумя путями:
1)жирные кислоты транспортируются в крови с помощью альбуминов;
2)ТГ, ФЛ, ХС, ЭХС и д.р. липиды транспортируются в крови в составе липопротеинов.
Обмен липопротеинов
Липопротеины (ЛП) – это надмолекулярные комплексы сферической формы, состоящие из липидов, белков и углеводов. ЛП имеют гидрофильную оболочку и гидрофобное ядро. В гидрофильную оболочку входят белки и амфифильные липиды - ФЛ, ХС. В гидрофобное ядро входят гидрофобные липиды - ТГ, эфиры ХС и т.д. ЛП хорошо растворимы в воде.
В организме синтезируются несколько видов ЛП, они отличаются химическим составом, образуются в разных местах и осуществляют транспорт липидов в различных направлениях.
ЛП разделяют с помощью:
1)электрофореза, по заряду и размеру, на α-ЛП, β-ЛП, пре-β-ЛП и ХМ;
2)центрифугирования, по плотности, на ЛПВП, ЛПНП, ЛППП, ЛПОНП и ХМ.
84
Соотношение и количество ЛП в крови зависит от времени суток и от питания. В постаб-сорбтивный период и при голодании в крови присутствуют только ЛПНП и ЛПВП.
Основные виды липопротеинов
Состав, % |
ХМ |
ЛПОНП |
|
|
||
(пре-β-ЛП) ЛППП |
|
|
|
|
||
(пре-β-ЛП) ЛПНП |
|
|
|
|
||
(β-ЛП) |
ЛПВП |
|
|
|
|
|
(α-ЛП) |
|
|
|
|
|
|
Белки |
2 |
10 |
11 |
22 |
50 |
|
ФЛ |
3 |
18 |
23 |
21 |
27 |
|
ХС |
2 |
7 |
8 |
8 |
4 |
|
ЭХС |
3 |
10 |
30 |
42 |
16 |
|
ТГ |
85 |
55 |
26 |
7 |
3 |
|
Плотность, г/мл 0,92-0,98 0,96-1,00 0,96-1,00 1,00-1,06 |
1,06-1,21 |
||||
Диаметр, нм |
>120 30-100 |
30-100 |
21-100 |
7-15 |
|
Функции Транспорт к тканям экзоген-ных липидов пищи |
|
Транспорт к |
|||
тканям эндоген-ных липидов пе-чени |
Транспорт к тканям эндоген-ных |
липидов пе-чени Транспорт ХС |
|
в ткани |
Удаление из-бытка ХС |
|
|
|
из тканей |
|
|
|
|
Донор |
|
|
|
|
апо А, С, Е |
|
|
|
|
Место образо-вания энтероцит |
гепатоцит |
в крови из ЛПОНП |
в |
|
крови из ЛППП гепатоцит |
|
|
|
|
Апо В-48, С-II, Е В-100, С-II, Е |
В-100, Е |
В-100 А-I С-II, Е, D |
|
|
Норма в крови |
< 2,2 ммоль/л 0,9- 1,9 ммоль/л |
|
||
Апобелки |
|
|
|
|
85
Белки, входящие в состав ЛП, называются апопротеины (апобелки, апо). К наиболее рас-пространенным апопротеинам относят: апо А-I, А-II, В-48, В- 100, С-I, С-II, С-III, D, Е. Апо-белки могут быть периферическими (гидрофильные: А-II, С-II, Е) и интегральными (имеют гидрофобный участок: В-48, В-100). Периферические апо переходят между ЛП, а интеграль-ные – нет. Апопротеины выполняют несколько функций:
Апобелок Функция Место обра-зования |
Локализация |
|
|||
А-I |
Активатор ЛХАТ, образование ЭХС печень |
ЛПВП |
|
|
|
А-II |
Активатор ЛХАТ, образование ЭХС |
ЛПВП, ХМ |
|
|
|
В-48 |
Структурная (синтез ЛП), рецепторная (фаго-цитоз ЛП) |
энтероцит |
|||
|
ХМ |
|
|
|
|
В-100 |
Структурная (синтез ЛП), рецепторная (фаго-цитоз ЛП) |
печень |
|||
|
ЛПОНП, ЛППП, ЛПНП |
|
|
|
|
С-I |
Активатор ЛХАТ, образование ЭХС Печень |
ЛПВП, ЛПОНП |
|||
С-II |
Активатор ЛПЛ, стимулирует гидролиз ТГ в ЛП Печень |
ЛПВП → ХМ, |
|||
ЛПОНП |
|
|
|
|
|
С-III |
Ингибитор ЛПЛ, ингибирует гидролиз ТГ в ЛП |
Печень |
ЛПВП → ХМ, |
||
ЛПОНП |
|
|
|
|
|
D |
Перенос эфиров холестерина (БПЭХ) |
Печень |
ЛПВП |
|
|
Е |
Рецепторная, фагоцитоз ЛП печень |
ЛПВП → ХМ, ЛПОНП, ЛППП |
|||
Ферменты транспорта липидов
Липопротеинлипаза (ЛПЛ) (КФ 3.1.1.34, ген LPL, около 40 дефектных аллелей) связана с гепарансульфатом, находящимся на поверхности эндотелиальных клеток капилляров крове-носных сосудов. Она гидролизует ТГ в составе ЛП до глицерина и 3 жирных кислот. При по-тере ТГ, ХМ превращаются в остаточные ХМ, а ЛПОНП повышают свою плотность до ЛППП и ЛПНП.
Апо С-II ЛП активирует ЛПЛ, а фосфолипиды ЛП участвуют в связывании ЛПЛ с по-верхностью ЛП. Синтез ЛПЛ индуцируется инсулином. Апо С-III ингибирует ЛПЛ.
ЛПЛ синтезируется в клетках многих тканей: жировой, мышечной, в легких, селезёнке, клетках лактирующей молочной железы. Ее нет в печени. Изоферменты ЛПЛ разных тканей отличаются по значением Кm. В жировой ткани ЛПЛ имеет Кm в 10 раз больше, чем в мио-карде, поэтому в жировая
86
ткань поглощает жирные кислоты только при избытке ТГ в крови, а миокард
– постоянно, даже при низкой концентрации ТГ в крови. Жирные кислоты в адипо-цитах используются для синтеза ТГ, в миокарде как источник энергии.
Печёночная липаза находиться на поверхности гепатоцитов, она не действует на зрелые ХМ, а гидролизует ТГ в ЛППП.
Лецитин: холестерол-ацил-трансфераза (ЛХАТ) находиться в ЛПВП, она переносит ацил с лецитина на ХС с образование ЭХС и лизолецитина. Ее активируют апо А-I, А-II и С-I.
лецитин + ХС → лизолецитин + ЭХС
ЭХС погружается в ядро ЛПВП или переноситься с участием апо D на другие ЛП.
Рецепторы транспорта липидов
Рецептор ЛПНП — сложный белок, состоящий из 5 доменов и содержащий углеводную часть. Рецептор ЛПНП имеет лиганды к белкам ano B-100 и апо Е, хорошо связывает ЛПНП, хуже ЛППП, ЛПОНП, остаточные ХМ, содержащие эти апо.
ЛПНП-рецептор синтезируется практически во всех ядерных клетках организма. Актива-ция или ингибирование транскрипции белка регулируется уровнем холестерина в клетке. При недостатке холестерина клетка инициирует синтез ЛПНП-рецептора, а при избытке — наоборот, блокирует его.
Стимулируют синтез рецепторов ЛПНП гормоны: инсулин и трийодтиронин (Т3), поло-вые гормоны, а глюкокортикоиды – уменьшают.
За открытие этого важнейшего рецептора липидного метаболизма Майкл Браун и Джозеф Голдштейн получили Нобелевскую премию по физиологии и медицине в 1985 году.
Белок, сходным с рецептором ЛПНП на поверхности клеток многих органов (печени, мозга, плаценты) имеется другой тип рецептора, называемый «белком, сходным с рецептором ЛПНП». Этот рецептор взаимодействует с апо Е и захватывает ремнантные (остаточные) ХМ и ЛППП. Так как ремнантные частицы содержат ХС, этот тип рецепторов также обеспечивает поступление его в ткани.
87
Кроме поступления ХС в ткани путём эндоцитоза ЛП, некоторое количество ХС поступа-ет в клетки путём диффузии из ЛПНП и других ЛП при их контакте с мембранами клеток.
В крови в норме концентрация:
•ЛПНП < 2,2 ммоль/л,
•ЛПВП > 1,2 ммоль/л
•общих липидов 4-8г/л,
•ХС < 5,0 ммоль/л,
•ТГ < 1,7 ммоль/л,
•Свободных жирных кислот 400-800 мкмоль/л
ОБМЕН ХИЛОМИКРОНОВ
Липиды, ресинтезированные в энтероцитах, транспортируется тканям в составе ХМ.
Образование ХМ начинается с синтеза апо В-48 на рибосомах. Апо В-48 и В-100 имеют общий ген. Если с гена копируется на мРНК только 48% информации, то с нее синтезируется апо В-48, если 100% - то с нее синтезируется апо В-100.
С рибосом апо В-48 поступает в просвет ЭПР, где он гликозилируется. Затем в аппарате Гольджи апо В-48 окружается липидами и происходит формирование «незрелых», насцентных ХМ.
Экзоцитозом насцентные ХМ выделяются в межклеточное пространство, поступают в лимфатические капилляры и по лимфатической системе, через главный грудной лимфатический проток попадают в кровь.
В лимфе и крови с ЛПВП на насцентные ХМ переносятся апо Е и С-II, ХМ превращаются в «зрелые». ХМ имеют довольно большой размер, поэтому они придают плазме крови опалесцирующий, похожий на молоко, вид. Под действием ЛПЛ ТГ ХМ гидролизуются на жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткань, а глицерол транспортируется с кровью в печень.
88
Когда в ХМ количество ТГ снижается на 90%, они уменьшаются в размерах, а апо С-II переносится обратно на ЛПВП, «зрелые» ХМ превращаются в «остаточные» ремнантные ХМ. Ремнантные ХМ содержат в себе фосфолипиды, холестерол, жирорастворимые витамины и апо В-48 и Е.
Через ЛПНП-рецептор (захват апо Е, В100, В48) ремнантные ХМ захватываются гепатоцитами. Путём эндоцитоза остаточные ХМ попадают внутрь клеток и перевариваются в лизосомах. ХМ исчезают из крови в течение нескольких часов.
ýí òåðî öè ò
н асцен тн ы й ХМ
ÕÌ
Â-48
ÔË, ÒÃ,
ÕÑ
Â-48
ñèí òåç ÕÌ
Î áì åí õè ë è ì è ê ðî í î â
|
|
|
|
|
|
|
|
|
Êðî âü |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
н асцен тн ы й ХМ |
|
|
|
|
|
|
|
|
|
|
||||||
Ëè ì ô à |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
ÕÌ |
Â-48 |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Å |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
ËÏ ÂÏ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
Ñ-II |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
зрелы й ХМ |
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
Å |
|
|
Î ðãàí û è òê àí è |
|||||||
ÕÌ |
|
|
|
|
|
|
|
|
ÕÌ Ñ-II |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
Â-48 |
|
|
|
|
|
|
|
|
|
|
|||
Â-48 |
|
|
|
|
|
|
|
|
|
|
|
ÒÃ |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
н асцен тн ы й |
|
|
|
|
|
|
|
|
|
ËÏ Ë |
в адип о цитах |
|||||||||||
ÕÌ |
|
|
|
|
|
|
|
|
|
ÆÊ |
|
|
|
ÆÊ |
|
|
ÑÎ 2 |
+ Í 2Î |
||||
ËÏ ÂÏ |
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
глицерин |
|
|
|
|
â ì û ø öàõ |
|||||||||||
|
|
|
|
|
|
Ñ-II |
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Ï å÷åí ü |
|
|||||||||
|
|
|
|
|
|
ðåì í àí òí û é ÕÌ |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
глицерин |
|
|||||||||||
|
|
|
|
|
|
ÕÌ |
Å |
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
рецеп то р к Е и В-48 |
||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Â-48 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Â-48 |
ô àãî öèòî ç |
Â-48 |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
ðåì í àí òí û é ÕÌ ÕÌ |
|
|
|
|
|
|
ÕÌ |
Å |
||||||||||||||
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Å |
ëèçî ñî ì àëüí î å |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
п еревариван ие |
|
||||||
ЖК, ХС, глицерин , АК
НАРУШЕНИЯ ОБМЕНА ХИЛОМИКРОНОВ
1. Абеталипопротеинемия (синдром Бассена-Корнцвейга)
При наследственном дефекте гена апо В — нарушается синтез апо В-100 в печени и апо В-48 в кишечнике. В результате в энтероцитах не формируются
89
ХМ, а в печени — ЛПОНП. В клетках этих органов накапливаются капли ТГ, нарушается всасывание пищевых липидов.
В крови наблюдается полное отсутствие ХМ, ЛПОНП, ЛППП и ЛПНП, уменьшение количества ХС, ФЛ, высших полиненасыщенных жирных кислот, Е и А витаминов.
Уже на первом месяце жизни отмечаются плохой аппетит, обильные испражнения с признаками стеатореи, гипотрофия. Развивается акантоцитоз (акантоциты - эритроциты с множественными шиловидными выростами), анемия, аритмия. Вследствие демиелинизации аксонов возникает прогрессирующая атаксия, нистагм, пигментная дегенерация сетчатки и отставание в умственном развитии.
Атаксия — это нарушение координации движений при поражении лобных долей головного мозна, мозжечка, путей глубокой чувствительности в спинном и головном мозге.
2. Семейная хиломикронемия ( гиперлипопротеинемии типа I) (менее 1%
всех случаев гиперлипопротеинемии)
Генетические дефекты ЛПЛ или апо С-II вызывают нарушение катаболизма ХМ, приводящий к гиперхиломикронемии. Высокий уровень ХМ сохраняется в плазме крови спустя 12 ч после приема пищи. За счет избытка ХМ плазма крови по виду напоминает молоко, при отстаивании ХМ образуют на ее поверхности сливкообразный слой. Хиломикронемия сопровождается гипертриглицеролемией - уровень ТГ в крови может превышать 11,3 ммоль/л. Содержание ХС остается нормальным или повышается (гиперхолестеринемия). Коэффициент ХС/ТГ менее 0,2. Развитие атеросклероза не характерно.
Семейная хиломикронемия возникает у детей в возрасте до 10 лет. У больных происходит отложение ТГ в коже и сухожилиях в виде ксантом. Эруптивные ксантомы могут занимать большую часть поверхности кожи.
ТГ также откладываются в печени, селезенке, поджелудочной железе и других органах, что вызывает в этих органах сужение просвета сосудов, уменьшение кровотока, развитие тромбозов и ишемических некрозов. Развивается панкреатит, гепато- и спленомегалия.
У пациентов нарушается память, возникают абдоминальные боли, тошнота, рвота, желтушность кожи, липемия роговицы. Прогрессирование панкреатита часто бывает причиной смерти больных.
90
ОБМЕН β-ЛИПОПРОТЕИНОВ
В промежутках между приемами пищи и при голодании необходимые для тканей липиды синтезируются преимущественно в печени. Печень — основной орган, где идёт синтез жирных кислот, ХС, ФЛ из продуктов гликолиза. Скорость синтеза липидов в печени существенно зависит от состава пищи. Если в пище содержится более 10% липидов, то скорость синтеза липидов в печени резко снижается.
Транспорт липидов из печени осуществляется с участием ЛПОНП. Синтез ЛПОНП идет также как и ХМ. Сначала на рибосомах синтезируется апо В-100, который потом в аппарате Гольджи соединяется с липидами. Так как апо В- 100 очень «длинный» белок (11536 АК), он покрывает поверхность всего ЛП.
После секреции ЛПОНП из печени в кровь, на них с ЛПВП переходят апо С-II и апо Е. Апо С-II активирует ЛПЛ, которая гидролизует ТГ ЛПОНП до жирных кислот и глицерина. Глицерол с кровью транспортируется в печень, а жирные кислоты – в ткань. Параллельно с потерей ТГ, ЛПОНП получают от ЛПВП ЭХС и ХС. В результате плотность ЛПОНП увеличивается, он превращается сначала в ЛППП, а затем в ЛПНП, при этом на ЛПВП возвращаются сначала апо С-II, а затем апо Е.
Содержание ЭХС и ХС в ЛППП достигает 45%; часть этих ЛП захватывается клетками печени через рецептор к ЛПНП (чувствителен к апо Е и апо В-100).
ЛПНП содержат до 55% ЭХС и ХС. ЛПНП являются основным поставщиком ХС в ткани. Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы к ЛПНП.
НАРУШЕНИЯ ОБМЕНА β-ЛИПОПРОТЕИНОВ
1. Семейная β-липопротеинемия ( гиперлипопротеинемия типа IIа) (10%
всех случаев гиперлипопротеинемии)
Возникает при дефекте апо В-100 (точечная мутация: арг замещен на глу), в результате чего уменьшается сродство ЛПНП к рецепторам В/Е, снижается катаболизм ЛПНП и увеличивается их концентрация в крови. β- липопротеинемия сопровождается гиперхолестеролемией, уровень ТГ в норме.
Семейный дефект апопротеида В-100 встречается с несколько меньшей частотой, чем семейная гиперхолестеринемия, и отличается от нее отсутствием сухожильных ксантом и более низким уровнем ХС.
91
2. Семейная гиперхолестеролемия (гиперлипопротеинемия типа IIа и IIв)
Наследственный дефект рецептора ЛПНП (к апо В/Е) (или белка апоВ-100) приводит к развитию распространённого наследственного заболевания — семейной гиперхолестеролемии.
При дефекте рецептора ЛПНП наблюдается триглицеролемия, при дефекте белка апоВ-100 – нет.
Угетерозигот (1:400) количество рецепторов ЛПНП на поверхности клеток снижено вдвое, а концентрация ХС в плазме примерно вдвое выше нормы (9-12 ммоль/л). ЛПНП фагоцитируются макрофагами. Нагруженные избытком ХС и других липидов, макрофаги откладываются в коже, сухожилиях и образуют ксантомы. Гиперхолестеролемия приводит к выраженному атеросклерозу, развитию ИБС и ранней смерти в результате инфаркта миокарда или инсульта.
Угомозигот (1:1000000) нет рецепторов к ЛПНП, концентрация ХС и ЛПНП в крови уже в раннем детском возрасте превышает норму в 5-6 раз (20-40 ммоль/л). Для пациентов характерно наличие не только ксантоматоза сухожилий, но и эруптивных ксантом на ягодицах, коленях, локтях, слизистой оболочке полости рта.
Избыток в крови ХС и ЛПНП способствует быстрому развитию атеросклероза и ИБС. Такие дети без экстренных мер лечения погибают в возрасте 5—6 лет.
ОБМЕН ЛПВП
ЛПВП выполняют 2 основные функции: они поставляют апо другим ЛП в крови и участвуют в так называемом «обратном транспорте ХС». ЛПВП синтезируются в печени и в небольшом количестве в тонком кишечнике в виде насцентных ЛПВП. Они имеют дисковидную форму, небольшой размер и содержат высокий процент белков и фосфолипидов. В печени в ЛПВП включаются апопротеины А, Е, С-II, ЛХАТ. В крови апо С-II и апо Е переносятся с ЛПВП на ХМ и ЛПОНП. насцентные ЛПВП практически не содержат ХС и ТГ и в крови обогащаются ХС, получая его из других ЛП и мембран клеток.
Для переноса ХС в ЛПВП существует сложный механизм. На поверхности ЛПВП находится фермент ЛХАТ — лецитин: холестерол-ацилтрансфераза. Этот фермент превращает ХС в ЭХС. Реакция активируется апо A-I, входящим в состав ЛПВП.
92