

изменений в иммунной системе, оптимизируют процессы регенерации при травматическом воспалении, уменьшают выраженность избыточ ных реакций (рис. 31–32). Поскольку при любой травме в ранние сро ки нарушается дифференцировка Т-лимфоцитов, в комплекс патогене тической терапии необходимо включить и препараты гормонов тиму са. В клинических условиях миелопид назначают внутримышечно по 3–6 мг 1 раз в день ежедневно в течение 5 дней, полиоксидоний – по 6 мг внутримышечно 2 дня подряд, затем через день, на курс – 5 инъек ций. В поздний травматический период для оптимизации регенерации, предотвращения развития аутоиммунных реакций перспективен оте чественный препарат профеталь (созданный на основе человеческого эмбрионального белка альфа-фетопротеина под руководством акаде миков В.А. Черешнева и Н.В. Васильева, д.м.н. С.Ю. Родионова). Ме ханизм действия профеталя в патогенетической терапии проникающе го ранения глаза представлен на рис. 33.
Таким образом, при проникающем ранении глаза развиваются фазные изменения функций иммунной системы, как общие для травмы в целом, так и специфичные для повреждения этого иммунологически привилегированного органа. Для коррекции иммунных нарушений могут быть использованы отечественные иммуномодуляторы нового поколения. Разработанная нами экспериментально-биологическая мо дель проникающего ранения глаза перспективна для оценки эффек тивности новых лекарственных препаратов.
XXVI. ТИПОВЫЕ НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ ПРИ САХАРНОМ ДИАБЕТЕ
Общие закономерности регуляции метаболизма
Обмен веществ, или метаболизм, представляет собой совокуп ность ферментативных реакций, складывающихся из процессов ката болизма и анаболизма. В результате процессов катаболизма посту пающие извне пищевые вещества – углеводы, жиры и белки – расщеп ляются до конечных продуктов с извлечением энергии, которая акку мулируется в макроэргических соединениях, а также используется для теплопродукции. Либо из огромного разнообразия поступающих извне соединений образуется ограниченное число строительных блоков – моносахаридов, глицерина, жирных кислот, аминокислот, ограничен ный набор низкомолекулярных карбоновых кислот (таких как пируват, лактат, кислоты цикла Кребса и др.), которые затем используются для построения собственных макромолекул. При анаболизме, основным назначением которого является синтез собственных макромолекул,
282

ПРОНИКАЮЩЕЕ РАНЕНИЕ ГЛАЗА
РЕГУЛЯТОРНЫЕ СИСТЕМЫ ОРГАНИЗМА
нервная |
эндокринная |
иммунная |
|||
|
|
|
|
|
|
|
|
|
|
|
|
ПОЛИОКСИДОНИЙ
ТРАВМАТИЧЕСКИЕ ИЗМЕНЕНИЯ  СТРЕССОРНЫЕ ИЗМЕНЕНИЯ
СТРЕССОРНЫЕ ИЗМЕНЕНИЯ
|
|
|
|
УСИЛЕНИЕ |
ПРОЦЕССЫ РЕГЕНЕРАЦИИ |
|
СОХРАНЕНИЕ СУПРЕССИИ |
||
|
|
ПРОТИВОВОСПАЛИТЕЛЬНОГО |
||
ПРИ ТРАВМАТИЧЕСКОМ |
|
РЕАКЦИИ |
|
|
|
|
ДЕЙСТВИЯ |
||
ВОСПАЛЕНИИ |
|
ГИПЕРЧУВСТВИТЕЛЬНОСТИ |
|
|
|
|
ГЛЮКОКОРТИКОИДОВ |
||
|
|
ЗАМЕДЛЕННОГО ТИПА |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
УВЕЛИЧЕНИЕ ЧИСЛА |
|
ОТМЕНА РАННЕЙ |
|
УМЕНЬШЕНИЕ |
|
ФИБРОБЛАСТОВ, |
|
|||
|
|
ИММУНОДЕПРЕССИИ |
||||
ВОСПАЛИТЕЛЬНОЙ |
|
БОЛЕЕ КОМПАКТНАЯ |
|
|||
|
|
АНТИТЕЛООБРАЗОВАНИЯ |
||||
КЛЕТОЧНОЙ |
|
И УПОРЯДОЧЕННАЯ |
|
|||
ИНФИЛЬТРАЦИИ |
|
СТРУКТУРА |
|
|
||
|
|
|
||||
|
|
|
РУБЦА |
|
|
|
|
|
|
|
МОДУЛЯЦИЯ ФУНКЦИЙ |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
ФАГОЦИТИРУЮЩИХ |
|
УСКОРЕНИЕ РЕГЕНЕРАЦИИ |
|
|
КЛЕТОК |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ПОВЫШЕНИЕ
ЧУВСТВИТЕЛЬНОСТИ ФАГОЦИТИРУЮЩИХ КЛЕТОК К β-АДРЕНЕРГИЧЕСКОЙ РЕГУЛЯЦИИ
ОПТИМИЗАЦИЯ ТЕЧЕНИЯ ВОСПАЛЕНИЯ
Рис. 31. Механизм действия полиоксидония в патогенетической терапии проникающего ранения глаза
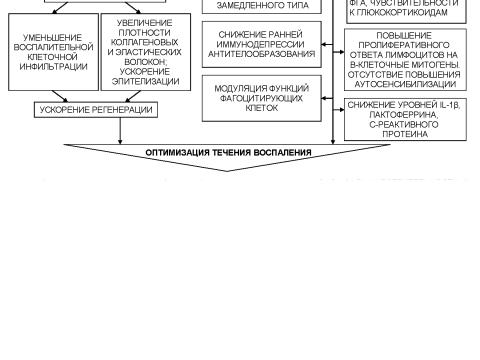
Рис. 32. Механизм действия миелопида при проникающем ранении глаза
|
|
|
|
|
|
|
ПРОНИКАЮЩЕЕ РАНЕНИЕ ГЛАЗА |
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
РЕГУЛЯТОРНЫЕ СИСТЕМЫ ОРГАНИЗМА |
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
нервная |
|
|
эндокринная |
|
иммунная |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
ПРОФЕТАЛЬ |
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
ТРАВМАТИЧЕСКИЕ ИЗМЕНЕНИЯ |
|
|
|
|
|
|
СТРЕССОРНЫЕ ИЗМЕНЕНИЯ |
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
СТИМУЛЯЦИЯ |
|
|
СНИЖЕНИЕ |
|
|
ТОРМОЖЕНИЕ ПОЗДНЕЙ |
|
|
АКТИВАЦИЯ ФАГОЦИТАРНОЙ |
|||||||||||||
ПРОЛИФЕРАЦИИ КЛЕ- |
|
ФОРМИРОВАНИЯ |
|
|
СТИМУЛЯЦИИ РЕАКЦИИ |
|
|
АКТИВНОСТИ МОНОЦИТОВ |
||||||||||||||
ТОК МНОГОСЛОЙНОГО |
|
ГРУБОЙ РУБЦОВОЙ |
|
|
ГИПЕРЧУВСТВИТЕЛЬНОСТИ |
|
|
ПЕРИФЕРИЧЕСКОЙ КРОВИ |
||||||||||||||
ПЛОСКОГО ЭПИТЕЛИЯ |
|
|
ТКАНИ |
|
|
ЗАМЕДЛЕННОГО ТИПА |
|
|
И ПЕРИТОНЕАЛЬНЫХ |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
МАКРОФАГОВ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
УСКОРЕНИЕ ЭПИТЕЛИЗАЦИИ |
|
|
|
ТОРМОЖЕНИЕ ПОЗДНЕЙ |
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
АКТИВАЦИЯ NK-КЛЕТОК |
||||||||||||||
|
|
ЗОНЫ ПОВРЕЖДЕНИЯ |
|
|
|
|
|
СТИМУЛЯЦИИ |
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
АНТИТЕЛООБРАЗОВАНИЯ |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
ОПТИМИЗАЦИЯ |
|
|
|
|
|
|
|
|
|
|
|
|
АКТИВАЦИЯ СОЗРЕВАНИЯ |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
РЕПАРАТИВНОЙ РЕГЕНЕРАЦИИ |
|
|
|
СНИЖЕНИЕ УРОВНЯ |
|
|
|
ДЕНДРИТНЫХ КЛЕТОК |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
АНТИТЕЛ К АНТИГЕНАМ |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ГЛАЗА |
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
ОПТИМИЗАЦИЯ ТЕЧЕНИЯ ВОСПАЛЕНИЯ |
|
|
|
|||||||||||||
Рис. 33. Механизм действия профеталя в патогенетической терапии проникающего ранения глаза
используются не только энергия, полученная в ходе катаболизма, но и указанные строительные блоки.
Разделение отдельных метаболических путей на процессы углеводного, липидного, аминокислотного и др. обменов весьма ус ловно, так как в организме они тесно взаимосвязаны как на уровне общего энергообеспечения клетки, так и на уровне общих ключевых метаболитов. Взаимосвязь на уровне общего энергообеспечения пред полагает конкуренцию разных метаболических путей за источники энергии. Поэтому, если в организме происходит, например, синтез триацилгицеролов в жировой ткани, этот процесс параллельно сопро вождается окислением в той же ткани глюкозы, которая обеспечивает синтез жира как энергетически за счет образования АТФ и НАДФН+H+, так и пластически – за счет образования ацетил-КоА. Таких примеров можно приводить множество. Но главная особенность
– это тесная координация различных метаболических путей в зависи мости от конкретного физиологического состояния организма.
Координация разных метаболических путей в организме носит многоуровневый характер. На уровне отдельных клеток, тканей и ор ганов регуляция отдельных метаболических путей осуществляется, во первых, по механизму положительных и отрицательных обратных свя зей метаболитами, которые могут выступать в роли аллостерических активаторов или ингибиторов отдельных ключевых ферментов, либо конкурентных ингибиторов – механизмы, хорошо изложенные в кур сах биохимии; во-вторых, наш организм унаследовал и такие филоге нетически древние механизмы регуляции, играющие очень важную роль у бактерий, как регуляцию на генетическом уровне (или на уров не транскрипции) синтеза тех или иных ферментов метаболитами. Пример – активация в печени синтеза глюкуронилтрансферазы, фер ментов микросомального окисления как эндогенными токсическими веществами, так и экзогенными – этанолом, барбитуратами, многими лекарственными препаратами. Однако на уровне целого организма ведущая роль принадлежит нейроэндокринным механизмам. Гормоны участвуют в регуляции метаболических путей, во-первых, за счет из менения активности отдельных ферментов, во-вторых, за счет регуля ции их синтеза на генетическом уровне, в-третьих, за счет изменения проницаемости клеточных мембран для ключевых метаболитов угле водного, жирового и аминокислотного обменов. Эти механизмы дей ствия гормонов во многом реализуются через внутриклеточные сиг нальные пути, связанные с образованием циклических нуклеотидов, инозитолтрифосфата, диацилглицерола и других посредников, в ре
286
зультате чего образуются активные протеинкиназы, которые фосфори лируют различные белки. В качестве последних могут выступать фер менты (ковалентная активация или ингибирование ферментов за счет присоединения фосфорной кислоты), ядерные факторы и регулятор ные белки хромосом (изменения биосинтеза белков и ферментов на генетическом уровне), белки рибосом (изменение биосинтеза белка на уровне трансляции), белки мембран (изменения проницаемости кле точных мембран для ключевых метаболитов углеводного, жирового, аминокислотного обменов).
Следует подчеркнуть, что сами ключевые метаболиты активно участвуют в регуляции обмена веществ по принципу обратных связей на многих уровнях нейроэндокринной системы. К таким ключевым метаболитам, концентрация которых поддерживается в узких гомео статических пределах, относится в углеводном обмене глюкоза крови (концентрация натощак – 3,33–5,55 ммоль/л). Внутриклеточно она всегда представлена в виде другого ключевого или центрального ме таболита всего углеводного обмена – глюкозо-6-фосфата. Если рас сматривать регуляцию обмена веществ с позиций функциональной системы П. К. Анохина, то именно уровень глюкозы в периферической крови – это наиболее важный сигнал, изменяющий по принципу об ратной афферентации весь метаболизм. Действие и восприятие этого сигнала в нейроэндокринной системе носит многоуровневый характер.
Во-первых, повышение концентрации глюкозы приводит не посредственно на уровне -клеток поджелудочной железы к усилению синтеза и секреции инсулина (рис. 34). Этот эффект связывают, преж де всего, с регуляцией глюкозой экзоцитоза содержащих инсулин гра нул β-клеток. Глюкоза проникает через наружную мембрану β-клеток с помощью белка GLUT2, транспортирующего глюкозу. У мышей с генетическим дефектом GLUT2 развивается гипергликемия и гипоин сулинемия, сопровождающаяся нарушением первой фазы секреции инсулина. Степень выраженности секреции инсулина β-клетками на ходится в прямой зависимости от уровня метаболизма в них глюкозы. Лимитирующим ферментом скорости катаболизма глюкозы в клетках является глюкокиназа, катализирующая превращение глюкозы в глюкозо-6-фосфат (во многих других клетках эта реакция катализи руется преимущественно гексокиназой). Доказано, что выраженность экзоцитоза гранул и высвобождения инсулина определяется концен трацией в -клетках промежуточных метаболитов обмена глюкозы, в частности пирувата и фосфоенолпирувата (метаболиты гликолиза), а также отношениями концентраций АТФ/АДФ и НАДФH+H+/НАДФ+.
287

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ca2+ |
|
|
|
|
|
|
Аргинин |
|
KАТФ- |
|
|
|
|
|
|
|
|
|
|
|
|
Потенциал- |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
каналы |
|
|
|
|
|
|
|
|
|
|
зависимые |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ca2+-каналы |
|
||
Лейцин |
|
|
|
|
|
|
|
|
|
|
-K+ |
Vm |
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
+ |
|
Глюкагон, GLP1, GIP |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
НАДФ+ |
- |
|
|
|
Gs АЦ |
|
Симпатическая |
||||||||||||||
|
|
|
|
|
|
|
|
|
нервнаясистема |
|||||||||||||||||
|
|
|
Пентозный |
- |
|
+ Ca |
2+ цАМФ |
|
Катехоламины |
|||||||||||||||||
GLUT2 |
цикл |
|
|
|
|
|
|
|
|
|
|
|
Gi АЦ |
|
Парасимпатическая |
|||||||||||
|
|
|
|
|
|
|
|
|
|
PKA |
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
цАМФ |
нервнаясистема |
|||||||||
Глюкоза |
|
|
Гл6ф |
НАДФН+Н |
|
|
|
|
|
|
|
|
Ацетилхолин |
|||||||||||||
|
|
|
АТФ/АДФ |
Ca2+ |
|
|
IP3 |
|
||||||||||||||||||
|
|
|
|
|
PIP2 |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
Пируват |
|
|
|
|
|
|
|
|
|
|
|
|
|
PLC |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
Ацетил-КоА |
|
|
|
PKC |
|
|
DAG |
PI3K |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
ЦиклКребса |
|
|
|
|
|
|
|
|
|
|
|
|
|
IRS |
Рецепторк |
|||||||
|
|
|
|
Тканевое дыхание |
|
|
|
|
|
|
|
|
|
инсулину |
||||||||||||
|
|
|
|
Окислительное |
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|||||||||
|
|
|
|
фосфорилирование |
|
|
|
|
|
Akt |
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Инсулин |
||||||
Митохондрии
 Экзоцитозивысвобождение инсулинаизклетки
Экзоцитозивысвобождение инсулинаизклетки
Рис. 34. Регуляция синтеза и секреции инсулина -клетками поджелудочной железы:
Гл6ф – глюкозо-6-фосфат, АЦ – аденилатциклаза, цАМФ – циклический АМФ, Vm – деполяризация или гипополяризация мембраны, Gs – G-протеин, активирующий адени латциклазу, Gi – G-протеин, ингибирующий аденилатциклазу, – повышение уровня,
– снижение уровня, PKA – протеинкиназа А, PKC – протеинкиназа C, PIP2 – фосфатиди линозитол-4,5-дифосфат, IP3 – инозитол-1,4,5-трифосфат, DAG – диацилглицерол, PLC – фосфолипаза C, PI3K – фосфатидилинозитол-3-киназа, IRS – субстрат рецептора к инсу лину (англ. insulin receptor substrate), Akt – `Akt-киназа (синоним: протеинкиназа B), GLUT2 – белок, транспортирующий глюкозу (англ. glucose transporter 2), GLP1 – глюка гоноподобный пептид-1 (англ. glucagon-like peptide-1; синоним: энтероглюкагон), GIP – глюкозозависимый инсулинотропный пептид (синонимы: гастроингибиторный пептид, гастроингибиторный полипептид), пунктирные линии без точки – закрытие каналов, пунктирные линии с точкой – активация процесса или открытие каналов, сплошные линии – последовательность реакций метаболизма и сигнальных путей
288
Для поиска новых антидиабетических препаратов существенны иссле дования последних лет роли в высвобождении инсулина -клетками пируват-цитратного, пируват-малатного и/или пируват-изоцитратного циклов, а также участия длинноцепочечных жирных кислот в качестве эндогенных лигандов ядерных рецепторов PPARs (англ. peroxisome- proliferator-activated receptors), которые на внутриклеточном уровне функционируют как сенсоры в регуляции обмена липидов на уровне изменения экспрессии генов. Показано, что увеличение уровня АТФ приводит к закрытию АТФ-зависимых SUR1/Kir6.2 калиевых каналов (KАТФ-каналов), что ведет к деполяризации мембраны. Деполяризация вызывает открытие потенциал-зависимых кальциевых каналов, что приводит к повышению проникновения Ca2+ в клетку. Увеличение уровня Ca2+ в клетке активирует фосфолипазу C, которая расщепляет фосфатидилинозитол-4,5-дифосфат на инозитол-1,4,5-трифосфат и диацилглицерол. Инозитолтрифосфат связывается с рецепторными белками эндоплазматического ретикулума. Это приводит к высвобож дению связанного внутриклеточного Ca2+ и резкому повышению его концентрации. Значительное увеличение концентрации в клетке Ca2+ приводит к высвобождению заранее синтезированного инсулина, хра нящегося в секреторных гранулах. Увеличение уровня НАДФH+H+, образующегося в пентозном цикле, приводит как к прямому увеличе нию экзоцитоза содержащих инсулин гранул, так и к усилению Ca2+- и АТФ-зависимого ответа за счет удлинения времени инактивации по тенциал-зависимых K+-каналов, реполяризующих клеточную мембра ну после ее деполяризации закрытием KАТФ-каналов. Гормоны и ме диаторы, повышающие уровень внутриклеточного циклического АМФ (цАМФ), также повышают секрецию инсулина β-клетками. Увеличе ние концентрации цАМФ приводит к повышению уровня внутрикле точного Ca2+ как непосредственно через активацию L-типа кальциевых каналов, так и через активацию протеинкиназы А. Последняя фосфо рилирует и закрывает KАТФ-каналы, что приводит к деполяризации плазматической мембраны. Помимо этого цАМФ повышает чувстви тельность внутриклеточных компонентов, ответственных за секрецию инсулина, к уровню внутриклеточного Ca2+, в результате чего Ca2+ индуцированная секреция усиливается при более низкой концентрации Ca2+. Протеинкиназа А также быстро фосфорилирует белки, ответст венные за секрецию инсулина. Наконец, цАМФ стимулирует экспрес сию инсулиновых генов как непосредственно за счет связывания с цАМФ-отвечающими элементами или CRE (англ. cAMP response elements) промотора инсулинового гена, так и опосредованно – за счет фосфорилирования транскрипционного фактора CREB (англ. cAMP
289
response element-binding protein – протеин, связывающийся с CRE
последовательностями ДНК).
Во-вторых, от уровня глюкозы прямо зависит активность глю корецепторов в гипоталамусе. По сути, глюкорецепторы – это важ нейшее афферентное звено нейроэндокринной регуляции обмена не только углеводов, но и липидов и аминокислот. Центральное звено регуляции представлено нейронами вентромедиального и латерально го гипоталамуса и тесно связанным с последними лимбико ретикулярным комплексом. Нейроны вентромедиального гипоталаму са формируют центр насыщения, именно в них находятся глюкорецеп торы. Повреждение этих нейронов ауротиоглюкозой приводит в экс перименте к повышению потребления пищи и ожирению. Возрастное повышение порога чувствительности этих нейронов к глюкозе доказа но и у людей, страдающих ожирением и диабетом 2-го типа. Напро тив, экспериментальное повреждение латеральных ядер гипоталамуса приводит к отказу от приема пищи. Указанные центры, с одной сторо ны, участвуют в формировании чувства голода и мотивации к приему пищи, с другой – через сегментарные отделы вегетативной нервной системы и эндокринную систему выступают как эфферентное звено регуляции в изменении углеводного, жирового и аминокислотного обменов. Парасимпатическая нервная система повышает секрецию
инсулина -клетками и тормозит секрецию глюкагона -клетками ост ровков Лангенгарса, а симпатическая (через α2-адренорецепторы) ока зывает прямо противоположный эффект. Активация α2 адренорецепторов приводит к гиперполяризации мембран β-клеток за счет открытия KАТФ-каналов, что предупреждает открытие потенциал зависимых кальциевых каналов и повышение уровня кальция с после дующим экзоцитозом секреторных гранул.
Из всех гормонов только инсулин снижает уровень глюкозы, что не случайно, так как именно гипогликемия наиболее опасна для организма, вызывая необратимые изменения в нейронах центральной нервной системы. В повышении же уровня глюкозы участвуют:
глюкагон, соматотропный гормон (СТГ) – основные гормо ны, продукция которых изменяется в условиях голода и связанной с ним гипогликемии;
адреналин, глюкокортикоиды – главные гормоны стресса (мобилизация глюкозы повышает на энергетическом уровне рези стентность организма к стрессорам);
тироксин – основной гормон, регулирующий энергетиче ский обмен (повышает уровень глюкозы в основном опосредованно,
290
через повышение чувствительности тканей к катехоламинам).
В норме существует суточная периодичность секреции ука занных гормонов. В дневное время, в основном, секретируется инсу лин, что обусловлено приемами пищи, а ночью при правильном режи ме питания – СТГ и глюкагон, что связано с уменьшением уровня глюкозы (народная пословица гласит: «Дети растут во время сна», – что подтверждается много раз доказанным научным фактом повыше ния продукции СТГ в ночное время). Поэтому в дневное время как основной источник энергии тканями потребляется глюкоза, так как основная направленность действия инсулина – это увеличение исполь зования глюкозы тканями в дихотомическом и апотомическом путях, печенью и мышцами для синтеза гликогена, а жировой тканью для синтеза жира. Напротив, в ночное время и при голодании, когда пре имущественно секретируются контринсулярные гормоны, основной источник энергии – неэтерифицированные жирные кислоты (НЭЖК), так как все вышеназванные контринсулярные гормоны обладают жи ромобилизующим действием, т.е. биоэнергетика переключается с ка таболизма углеводов на катаболизм липидов. Эти биоритмы сущест венно нарушаются при стрессе или при переедании в вечернее время, что способствует развитию ожирения (народная пословица: «Плотный ужин пожелай врагу») и диабета 2-го типа.
Итак, другим ключевым метаболитом (но уже жирового обме на) являются НЭЖК, образующиеся в основном за счет распада триа цилглицеролов в жировой ткани. Уровень НЭЖК существенно повы шается за счет активации триацилглицероллипазы жировой ткани кон тринсулярными гормонами – глюкагоном, СТГ, адреналином, про стагландинами, адренокортикотропным гормоном (АКТГ). В свою очередь, НЭЖК существенно снижают проницаемость клеточных мем бран для глюкозы и за счет этого препятствуют снижению ее уровня при голодании. Этот эффект имеет очень важное значение, так как запасы гликогена в печени в отличие от запасов жира в жировой ткани очень ограничены. С другой стороны, именно избыточное повышение НЭЖК под действием СТГ может приводить к гипергликемии.
К другим важнейшим метаболитам, регулирующим секрецию инсулина и контринсулярных гормонов, относятся аминокислоты (особенно аланин, концентрация которого резко возрастает при голо дании, а также аргинин и лейцин). Аргинин и лейцин стимулируют секрецию как инсулина, так и глюкагона, в то время как аланин изби рательно стимулирует секрецию глюкагона, но не инсулина. Эта ами нокислота является основным источником для синтеза глюкозы при голодании за счет активации глюконеогенеза.
291
Указанными соединениями и механизмами отнюдь не ограни чивается регуляция метаболизма. Доказана, например, важная роль изменений концентрации самого инсулина для регуляции активности пищевых центров гипоталамуса.
Известно, что энтеральное поступление глюкозы приводит к значительно более высокой секреции инсулина, чем парентеральное. Оказалось, что это связано с продукцией клетками желудочно кишечного тракта глюкагона, гастрина, секретина и холецистокинина (ранее имел название панкреозимин). Вазоактивный интестинальный пептид (VIP) стимулирует секрецию как инсулина, так и глюкагона, что важно для изменения их продукции при всасывании продуктов пищеварения. Помимо этого, секреция инсулина стимулируется глю козозависимым инсулинотропным полипептидом (GIP; синонимы: глюкозозависимый инсулинотропный пептид, гастроингибиторный пептид, гастроингибиторный полипептид, желудочный ингибиторный пептид; состоит из 42 аминокислотных остатков, часть аминокислот ной последовательности совпадает с секретином, а другая – с глюка гоном), вырабатываемым K-клетками слизистой оболочки двенадца типерстной и проксимальной части тощей кишок, а также пептидным гормоном из семейства секретина – глюкагоноподобным пептидом-1 (GLP-1, англ. glucagon-like peptide-1; синоним энтероглюкагон), секре тируемого L-клетками слизистой оболочки подвздошной и толстой кишок. Оба полипептида относятся к инкретинам, т.е. к гормонам, вырабатывающимся в кишечнике при приеме пищи и действующим через кровь. Стимуляторами их секреции являются жиры и углеводы (глюкоза), поступающие в кишечник с переваренной в желудке пищей. При этом показано, что глюкоза, введенная внутривенно, не влияет на продукцию GLP-1. Как и GLP-1, GIP и глюкагон стимулируют про дукцию инсулина через повышение уровня цАМФ (см. рис. 34).
Продукцию инсулина и глюкагона ингибирует соматостатин (секретируется -клетками островков Лангенгарса).
Важнейшую роль в регуляции энергетического метаболизма играют грелин, являющийся гормоном голода, и его функциональный антагонист – лептин. Грелин секретируется -клетками островков Лан герганса, а также P/D1-клетками слизистой оболочки фундального отдела желудка. Концентрация грелина перед приемом пищи увеличи вается, а после него уменьшается. Грелин взаимно дополняет цитокин лептин, производимый в жировой ткани и вызывающий чувство на сыщения. Грелин секретируется и в дугообразном ядре гипоталамуса, активируя чувство голода, где он через рецепторы, экспрессируемые
292

на нейронах в дугообразном ядре и вентромедиальном гипоталамусе, стимулирует секрецию СТГ передней долей гипофиза. Лептин – цито кин с системным эндокринным действием, регулирующий энергетиче ский обмен и массу тела. Основная роль – передача в гипоталамус информации о массе тела и жировом обмене. Оказывает анорексиген ное действие (подавляет аппетит). Является белком, состоящим из 167 аминокислот, секретируется адипоцитами (клетками жировой ткани). Лептин вызывает чувство насыщения, действуя на гипоталамус, бло кируя синтез и высвобождение нейропептида Y, вызывающего чувство голода. Врожденная недостаточностъ лептина у человека и лаборатор ных грызунов приводит к развитию тяжёлой формы ожирения. В фи зиологических условиях лептин угнетает синтез инсулина, а инсулин, воздействуя на жировую ткань, стимулирует продукцию лептина.
Сахарный диабет
В условиях патологии все эти процессы регуляции обмена могут существенно нарушаться. Наиболее ярко это проявляется при такой типовой патологии регуляции обмена веществ, как сахарный диабет. Для него характерны глубокие нарушения углеводного, жиро вого, белкового и аминокислотного, водно-электролитного обменов и их регуляции.
Сахарный диабет – это очень распространенная и социально значимая патология. По данным ВОЗ1,
347 млн чел. во всем мире страдают сахарным диабетом;
в 2004 г. 3,4 млн чел. умерли от последствий высокого со держания сахара в крови натощак, такое же число случаев смерти про изошло, по оценкам, и в 2010 г.;
более 80% случаев смерти от диабета происходит в странах с низким и средним уровнем дохода;
по прогнозам, в 2030 г. диабет станет седьмой по значимости причиной смерти;
благодаря здоровому питанию, регулярной физической ак тивности, поддержанию нормального веса тела и воздержанию от упо требления табака можно предотвратить или отсрочить заболевание диабетом 2-го типа.
Са́харный диабе́т (лат. diabetes mellitus) – группа эндокрин ных заболеваний, развивающихся вследствие абсолютной или относи тельной (нарушение взаимодействия с клетками-мишенями) недоста
1 Диабет. Информационный бюллетень N°312. Март 2013 г.
293
точности гормона инсулина; характеризуется нарушением вследствие этого всех видов обмена, и в первую очередь обмена углеводов.
В истории научного изучения сахарного диабета можно выде лить 3 этапа смены представлений о главных факторах его патогенеза.
Первый этап – концепция недержания воды. Первые описания этого патологического состояния выделяли самые яркие его симптомы
– потерю жидкости (полиурия) и неутолимую жажду (полидипсия). Термин «диабет» (лат. diabetes), который впервые был использован греческим врачом Эрасистратусом (Erasistratus, известным также как
Apollonius Memphites или Apollonius of Memphis; родился в Мемфисе в Египте в III в. до н. э.), происходит от др.-греч. διαβήτης, что перево дится как «проходить через» или как «сифон». Таково было представ ление о диабете как состоянии, при котором человек непрерывно теря ет жидкость и постоянно ее восполняет вследствие неспособности ее задерживать. Это один из основных симптомов диабета – полиурия (избыточное выделение мочи). Гален охарактеризовал данное заболе вание как мочевую диаррею (лат. diarrhea urinosa). Главным звеном патогенеза сахарного диабета считали утрату организмом способности удерживать жидкость.
Второй этап – концепция недержания глюкозы. В 1675 г. То мас Уиллис (Thomas Willis, 1621–1675) показал, что при полиурии (повышенном выделении мочи) моча может быть «сладкой», а может быть и «безвкусной». В первом случае он добавил к слову диабет (лат. diabetes) слово mellitus, что с латинского означает «сладкий, как мёд» (лат. diabetes mellitus), а во втором — «insipidus», что означает «без вкусный». Безвкусным был назван несахарный диабет – патология, вызванная либо заболеванием почек (нефрогенный несахарный диа бет), либо заболеванием гипофиза (нейрогипофиза) и характеризую щаяся нарушением секреции или биологического действия антидиуре тического гормона. Мэтью Добсон (Mathew Dobson, 1735–1784) дока зал, что сладкий вкус мочи и крови больных диабетом обусловлен большим содержанием глюкозы. Древние индийцы заметили, что моча больных диабетом притягивает муравьёв, и назвали это заболевание «болезнью сладкой мочи». Корейские, китайские и японские аналоги этого слова основываются на той же идеограмме и также означают «болезнь сладкой мочи». Индийские врачи Сушрута (Sushruta) и Чара ка (Charaka) в 400–500 г. н. э. указывали на существенные различия двух разных форм диабета: ювенильного (в настоящее время обозна чаемого как тип 1) и диабета тучных (рассматриваемого в настоящее время как тип 2).
294
Третий этап – концепция абсолютной и относительной недос таточности инсулина. Первые доказательства участия поджелудочной железы в патогенезе этого заболевания были получены О. Минков ским (Oskar Minkowski, 1858–1931) и И. Мерингом (Joseph Freiherr von Mering, 1849–1908) в 1889–1892 гг. Они создали первую эксперимен тальную модель сахарного диабета путем удаления у собак поджелу дочной железы. Позже, в 1892 г. О. Минковский пересадил проопери рованной собаке ее собственную поджелудочную железу и тем самым задержал развитие диабета. В 1889–1990 гг. русский учёный Леонид Васильевич Соболев в диссертации «К морфологии поджелудочной железы при перевязке её протока при диабете и некоторых других ус ловиях» показал, что перевязка выводного протока поджелудочной железы приводит к полной атрофии ацинозного (экзокринного) отде ла, тогда как панкреатические островки остаются нетронутыми. На основании опытов Л. В. Соболев пришёл к выводу: «функцией пан креатических островков является регуляция углеводного обмена в ор ганизме. Гибель панкреатических островков и выпадение этой функ ции вызывает болезненное состояние – сахарное мочеизнурение». Ис пользуя предложенную Л. В. Соболевым методику, в 1921 г. Бастинг
(Frederick Grant Banting, 1891–1941; Нобелевская Премия, 1923 г.) и Бест (Charles Herbert Best, 1899–1978) выделили из поджелудочной железы новорожденного теленка инсулин. В 1936 г. Гарольд Химсворт
(Harold Percival Himsworth, 1905–1993) выяснил, что существует диа бет 1-го и 2-го типов.
Большую роль в выяснении патогенеза диабета сыграла мо дель аллоксанового диабета. Однако в настоящее время из-за токсич ности аллоксана ее стали использовать относительно редко. Наиболее адекватной экспериментальной моделью индуцированного диабета 1 го типа считается стрептозотоциновый диабет (стрептозотоцин – ан
тибиотик, избирательно повреждающий -клетки у крыс, мышей и собак и обладающий малой токсичностью). Выведены и инбредные линии мышей и других животных с наследственным сахарным диабе том как 1-го, так и 2-го типов.
Классификация сахарного диабета
Единой общепринятой классификации сахарного диабета нет. По этиологии и патогенезу большинство исследователей выделяет следующие формы сахарного диабета.
I. Сахарный диабет 1-го типа. В основе лежит деструкция β клеток, которая приводит к абсолютной инсулиновой недостаточно
295
сти. Чаще развивается в детском возрасте. По механизму развития и этиологии выделяют следующие формы:
аутоиммунный;
идиопатический.
II. Сахарный диабет 2-го типа. Характеризуется наруше ниями секреции инсулина на фоне инсулинорезистентности (снижения ответа клеток на инсулин), вследствие чего развивается относительная инсулиновая недостаточность.
III. Гестационный сахарный диабет. Развивается в 2–5%
случаев от общего числа беременностей, характеризуется сочетанием неадекватной секреции инсулина и снижения ответа клеток на него. Примерно у 20–50% женщин, перенесших во время беременности эту
форму диабета, впоследствии развивается диабет 2-го типа.
IV. Другие типы диабета, развиваются при:
генетических дефектах функции β-клеток;
генетических дефектах рецепции инсулина и передачи внут риклеточных сигналов с инсулиновых рецепторов;
нарушениях в иммунной системе, например, латентный ау тоиммунный диабет взрослых (англ. LADA, latent autoimmune diabetes of adults);
болезнях экзокринной части поджелудочной железы;
эндокринопатиях;
под действием лекарственных препаратов и других химиче ских соединений;
при инфекциях;
генетических синдромах, сочетающихся с сахарным диабе
том.
Основные особенности сахарного диабета 1-го и 2-го типов представлены в табл. 13.
Этиология и патогенез сахарного диабета зависят от его типа, поэтому будут рассмотрены отдельно.
Сахарный диабет 1-го типа
Главным звеном патогенеза сахарного диабета 1-го типа явля ется недостаточная продукция инсулина -клетками поджелудочной железы (абсолютная инсулиновая недостаточность). Этот тип сахарно го диабета раньше обозначали как инсулинозависимый, ювенильный, или детский диабет. Поскольку больным этой формой заболевания, как правило, вводят с заместительной целью препараты инсулина, для оценки продукции собственного инсулина -клетками поджелудочной
296
Таблица 13
Сравнительная характеристика сахарного диабета 1-го и 2-го типов
Проявления |
Сахарный диабет |
|
|
1-го типа |
2-го типа |
Начало |
Внезапное |
Постепенное |
Возраст начала заболевания |
Чаще у детей |
Чаще у взрослых |
Состояние жировой ткани |
Исхудание или |
Чаще ожирение |
Кетоацидоз |
норма |
|
Часто |
Редко |
|
Аутоантитела |
Обычно |
Отсутствуют |
Уровень эндогенного |
выявляются |
|
Снижен |
Нормальный, |
|
инсулина (чаще оценивается |
или отсутствует |
сниженный или |
по концентрации С-пептида) |
|
повышенный |
Конкордантность у |
50% |
90% |
однояйцевых |
|
|
близнецов |
|
|
Частота |
Около 10% |
Около 90% |
железы важное значение имеет определение C-пептида. В эндокрин ных -клетках поджелудочной железы вначале синтезируется предше ственник гормона – проинсулин. Последний при секреции подвергает ся протеолитическому расщеплению с образованием инсулина и C пептида. Поэтому концентрация C-пептида в крови отражает интен сивность секреции собственного инсулина, что важно при оценке эн докринной функции поджелудочной железы при сахарном диабете в условиях инсулинотерапии (при определении концентрация инсулина в крови в этом случае будет выявляться как введенный извне инсулин, так и собственный).
Отправным моментом в развитии абсолютной недостаточно сти инсулина является массивное разрушение эндокринных -клеток поджелудочной железы (островков Лангерганса) и, как следствие, кри тическое снижение уровня инсулина в крови. Массовая гибель клеток поджелудочной железы может иметь место вследствие вирус ных инфекций, онкологических заболеваний, панкреатита, токсиче ских поражений поджелудочной железы, стрессовых состояний, ауто иммунных реакций, развивающихся по механизмам как гуморального, так и клеточноопосредованного иммунного ответа. В последнем слу
297
чае в качестве клеточных эффекторов выступают цитотоксические Т лимфоциты, активированные Th1-клетки и макрофаги. Этот тип диа бета характерен для детей и лиц молодого возраста (до 40 лет). У че ловека это заболевание зачастую является генетически детерминиро ванным и обусловленным дефектами ряда генов, расположенных в 6-й хромосоме. Эти дефекты формируют предрасположенность к аутоим мунной агрессии организма к клеткам поджелудочной железы и отри цательно сказываются на регенерационной способности β-клеток. Провоцирующими факторами могут являться длительная гипоксия клеток поджелудочной железы и высокоуглеводистая, богатая жирами и бедная белками диета, что приводит к снижению секреторной актив ности островковых клеток и впоследствии к их гибели. После начала массивной гибели клеток запускается механизм их аутоиммунного повреждения.
Экспертный комитет Американской Ассоциации диабета
(англ. ADA, the American Diabetes Association) в 2002 г. предложил этиологические критерии (табл. 14), на основании которых сахарный диабет 1-го типа подразделяют на тип 1A (иммуноопосредованный) и тип 1B (другие формы диабета с выраженной недостаточностью инсу лина). В начальной стадии развития диабета отличить диабет типа 1A от диабета 2-го типа не всегда простая задача. Наилучшим критерием для выявления диабета 1A типа является выявление специфических аутоантител. Положительный тест на аутоантитела с тест-системами, дающий менее 1 позитивного результата у 100 обследуемых кон трольной группы (т.е. со специфичностью ≥99%), с большой вероят ностью указывает на тип 1A диабета. Неиспаноговорящие белые дети обычно страдают диабетом типа 1A, тогда как взрослые старше 40 лет обычно имеют диабет 2-го типа. У более чем 90% таких детей выявля ется один из четырех типов аутоантител против островков Лангенгар са. Напротив, среди половины черных или латиноамериканских детей аутоантитела отсутствуют. У этих детей в 8–10 раз чаще развивается диабет 2-го типа в раннем возрасте, у многих из них есть такой фактор риска, как ожирение, но отсутствуют аллели HLA, характерные для типа 1A диабета. Ожирение не предупреждает от развития диабета типа 1А, хотя оно обычно ассоциировано с инсулинорезистентными формами диабета. Важно заметить, что пациенты, имеющие и рези стентность к инсулину и тип 1А диабета, могут иметь высокие уровни инсулина натощак и C-пептида, но сниженную стимулированную сек рецию инсулина.
298
Таблица 14
Основные критерии сахарного диабета IA типа, отличающие его отдругих типов диабета
|
Тест на аутоанти |
|
|
Тип диабета |
тела против ост |
Генетика |
Комментарии |
|
ровков Ланген |
|
|
|
гарса |
|
|
Тип IA |
Положителен |
HLA 30–50% |
90% неиспаноя |
|
>90% |
DR3 и DR4 |
зычных белых |
|
|
|
детей |
|
|
HLA 90% DR3 |
50% черных де |
|
|
или DR4 |
тей |
|
|
HLA <3% |
50% латиноаме |
|
|
DQB1*0602 |
|
Тип IB |
Отрицательный |
Неизвестна |
риканских детей |
Редко у белых |
|||
Тип II |
Отрицательный |
Неизвестна |
Если есть анти |
|
|
|
тела, вероятен |
|
|
|
латентный ауто |
|
|
|
иммунный диа |
|
|
|
бет взрослых |
|
|
|
(LADA), гапло |
|
|
|
тип HLA являет |
|
|
|
ся сходным с |
Другие |
Отрицательный |
Мутации |
типом IA |
|
|||
|
|
MODY (англ. |
|
|
|
maturity-onset |
|
|
|
diabetes of the |
|
|
|
young), другие |
|
|
|
синдромы |
|
Экспериментальные модели сахарного диабета 1-го типа
уживотных
1.Экспериментальные модели индуцированного сахарного диабета у животных. Наиболее старой моделью, использующейся с XIX в. по настоящее время, является хирургическая операция панкреа тэктомии. В настоящее время эта модель используется при разработке
299
методов трансплантации β-клеток островков Лангерганса у разных видов экспериментальных животных.
Одной из первых моделей, не связанных с хирургическим вмешательством, является аллоксановый диабет. Аллоксан (2,4,5,6 тетраоксипиримидин; 5,6-диоксиурацил) впервые описан Бругнателли
(Brugnatelli) в 1818 г. Вёхлер (Wöhler) и Лиебиг (Liebig) предложили название «аллоксан» при описании его химического синтеза окислени ем мочевой кислоты. Диабетогенное действие этого соединения было открыто много лет спустя (Dunn J.S., Sheehan H.L., McLethie N.G.B., 1943). Было установлено, что введение аллоксана кроликам приводит к развитию специфического некроза островков поджелудочной желе зы. С тех пор экспериментальная модель аллоксанового диабета стала широко использоваться для изучения патогенеза сахарного диабета 1 го типа.
Другой моделью является сахарный диабет, индуцируемый у крыс или мышей введением стрептозотоцина. Стрептозотоцин (2 деокси-2-(3-метил-3-нитрозоуреидо)-D-глюкопираноза) синтезируется Streptomycetes achromogenes и используется в медицине как химиоте рапевтический препарат для лечения злокачественных опухолей под желудочной железы, развивающихся из β-клеток островков Лангер ганса. В дозе 65 мг/кг массы тела у крыс и 190 мг/кг массы тела у мы шей при однократном внутрибрюшинном введении приводит к разви тию у 100% животных тяжелого диабета 1-го типа, связанного с пря мым повреждением β-клеток островков Лангерганса поджелудочной железы. По нашим данным, у крыс диабет протекает более тяжело, чем у мышей, и без введения инсулина приводит к 100% летальности крыс уже через 72 ч после инъекции стрептозотоцина в вышеуказан ной дозе вследствие развития гипергликемии и гиперкетонемии (Ши лов Ю.И. и др., 2013). Уровень глюкозы уже через 24 ч после введения стрептозотоцина крысам в среднем повышается с 5,50±0,15 в контроле до 27,88±2,54 ммоль/л (средняя арифметическая±стандартная ошибка средней; у отдельных животных он увеличивается в этот срок до 52 ммоль/л). Летальность у мышей при введении значительно более вы сокой дозы стрептозотоцина (190 мг/кг) в течение первых 10 суток эксперимента, по нашим данным, составляет 47,9% без введения инсу лина. Средняя концентрация глюкозы на 10-е сутки эксперимента у мышей с сахарным диабетом, по нашим данным, составляет 20,33±0,75, а в контроле – 3,98±0,29 ммоль/л. В течение первых трех суток после введения стрептозотоцина как у крыс, так и у мышей час то развивается выраженная гипогликемия (вплоть до гипогликемиче ской комы и смерти) вследствие массивного выброса эндогенного ин
300
сулина при повреждении β-клеток островков Лангерганса и их дегра нуляции. Для предупреждения развития летальной гипогликемии ре комендуется поить животных в течение этого срока 10% раствором глюкозы вместо воды. Хотя в литературе описаны экспериментальные модели с введением более низких доз стрептозотоцина, наш опыт по казывает, что действие этого антибиотика подчиняется закономерно сти «все или ничего», и при использовании более низких доз препара та даже в сочетании с обогащенной жирами диетой диабет не развива ется при наблюдении за животными в течение нескольких месяцев.
Как стрептозотоцин, так и аллоксан проникают в β-клетки островков Лангерганса через транспортер глюкозы GLUT2 и разру шают их через образование активных форм кислорода и свободных радикалов.
Описаны экспериментальные модели аутоиммунного диабета у инбредных линий крыс с аллелями MHC II класса RT1 B/Du, опреде ляющих генетическую предрасположенность к диабету, при введении им кополимера полиинозиновой и полицитидиловой кислот (поли ИЦ), усиливающего продукцию интерферона-α (IFN-α). Показано, что повышение продукции IFN-α при вирусных инфекциях может способ ствовать развитию аутоиммунных реакций против β-клеток и у людей
сопределенными гаплотипами HLA.
2.Экспериментальные модели спонтанного сахарного диабе-
та у животных. NOD-мыши (англ. nonobese diabetic mice – нетучные диабетические мыши) – одна из наиболее широко исследуемых в на
стоящее время экспериментальных моделей естественно развивающе гося сахарного диабета. Впервые эти животные были получены груп пой S. Makino с соавт. (1980) при скрещивании одной из двух подли ний CTS-мышей, характеризовавшейся развитием полиурии и глюко зурии. У полученных путем направленного скрещивания NOD-мышей симптомы сахарного диабета проявлялись в виде полиурии, полидип сии, гипергликемии, глюкозурии и гиперхолестеринемии. Патоморфо логически развивалась выраженная лимфоцитарная инфильтрация ост ровков Лангерганса, снижение их количества и размера. Подобно са харному диабету типа 1A у человека, при котором для развития ауто иммунного повреждения β-клеток важную роль играет генетическая предрасположенность, связанная с определенными гаплотипами HLA I и II классов, NOD-мыши имеют мутации, связанные с отсутствием молекул, кодируемых локусом I-E комплекса Н-2 (аналог HLA-DR у человека), или с необычной структурой молекул, кодируемых локусом I-A главного комплекса гистосовместимости (аналог HLA-DQ у чело века). Поскольку продукты этих локусов, являясь молекулами гисто
301
совместимости II класса (MHC II класса), участвуют в презентации «чужих» антигенных пептидов Т-хелперам, именно они определяют возможность развития аутоиммунных реакций. Показано, что замеще ние отсутствующего I-E локуса I-E трансгеном предупреждает разви тие сахарного диабета. Сходные результаты получены при замещении дефектного I-A локуса трансгенным I-A геном с другой последова тельностью нуклеотидов. Помимо MHC II класса более чем 15 других генетических локусов участвуют в развитии заболевания, поэтому предрасположенность NOD-мышей к развитию сахарного диабета яв ляется полигеномной.
Развитие сахарного диабета у NOD-мышей связано с развити ем инсулита, который впервые выявляется у них в возрасте 4–5 не дель. К 7-месячному возрасту сахарный диабет развивается примерно
у90% самок и только у 60% самцов. Характерна мононуклеарная ин фильтрация островков Лангерганса, которая связана с аутоиммунной атакой их клеток и которая в конечном итоге приводит к полному раз рушению островков. Инфильтрат содержит Т-лимфоциты, макрофаги, NK-клетки, В-лимфоциты. Развитие аутоиммунных реакций у NOD мышей может быть связано и с дефектами регуляторных Т лимфоцитов, что характерно и для сахарного диабета 1-го типа у чело века. В связи с этим NOD-мыши являются удобным объектом для раз работки новых подходов предупреждения аутоиммунных реакций при диабете 1-го типа. Доказано участие аутоиммунного компонента и в патогенезе повреждения β-клеток в поздние сроки развития стрептозо тоцинового и аллоксанового диабета.
Вотличие от NOD-мышей, предрасположенность которых к развитию сахарного диабета носит полигенный характер, у некоторых линий крыс она является олигогенной. Сахарный диабет, спонтанно развивающийся у крыс BB (от англ. biobreeding rats; впервые получе ны в 1974 г. в Bio Breeding Laboratory), также является моделью са харного диабета типа 1A человека. Крысы BB имеют аутосомную ре циссивную мутацию, которая приводит к развитию выраженной Т лимфопении. Олигогенной является и предрасположенность к диабету
укрыс линии LETL (англ. Long-Evans Tokushima Lean rats), которые подобно крысам BB имеют аллели RT1 B/Du (аллель MHC II класса) с мутацией гена Cbl-b, приводящей к нарушениям проведения Т
клеточных внутриклеточных сигналов.
Одной из подлиний крыс LETL является линия KDP. Сахар ный диабет у крыс KDP развивается в 3–4-месячном возрасте, частота развития заболевания – 70–80% как у самок, так и у самцов. Для этих крыс нехарактерно развитие лимфопении, они имеют общий с крыса
302
ми BB гаплотип молекул главного комплекса гистосовместимости II класса RT1 B/Du. При патоморфологическом исследовании через 120– 220 дней жизни у них выявляется развитие инсулита, а также лимфо цитарная инфильтрация щитовидной железы и почек. Помимо этого у крыс KDP экспрессируется не связанный с главным комплексом гис тосовместимости ген, названный Cblb (англ. casitas B-lineage lym phoma b) и кодирующий убиквитиновую лигазу, которая важна для костимуляции через CD28 активации Т-лимфоцитов. Однако связи аналогов этого гена с сахарным диабетом у человека не выявлено. Сходство частоты развития сахарного диабета у самцов и самок крыс KDP с заболеванием человека указывает на высокую перспективность этой относительно недавно найденной модели для изучения патогенеза сахарного диабета 1-го типа человека.
В 2004 г. выведена новая линия крыс (Lenzen S. et al., 2004) из конгенной линии LEW.1AR1 через спонтанную мутацию, которая обо значена как линия LEW.1AR1/Ztm-iddm. У всех крыс этой линии экс прессируется аллель MHC II класса RT1 B/Du, для них не характерно развитие лимфопении, но развивается повреждение островков Лангер ганса, сходное с аналогичным повреждением у крыс BB и KDP. Са харный диабет 1-го типа развивается у них в возрасте 2–3 месяцев, характеризуется низким уровнем инсулина, гипергликемией, глюкозу рией и кетонурией. Частота развития заболевания около 70% и у са мок, и у самцов. Характерна низкая летальность без терапии инсули ном. Для этих крыс не характерно увеличение аутоантител к глутамат декарбоксилазе и к тирозиновой фосфатазе ICA512 (обозначаемой также как IA-2). Иммуноциты, инфильтрирующие поджелудочную железу, не атакуют щитовидную железу и другие железы.
Сахарный диабет 2-го типа
Главным звеном патогенеза сахарного диабета 2-го типа явля ется нарушение ответа клеток тканей организма на инсулин (инсули норезистентность) как следствие изменения структуры или уменьше ния количества специфических рецепторов для инсулина, изменения структуры самого инсулина или нарушения внутриклеточных меха низмов передачи сигнала с рецепторов инсулина (рис. 35). Нарушение адекватного ответа на нормальные уровни инсулина мышечной и жи ровой ткани, печени приводит к развитию гипергликемии (мобилиза ция глюкозы печенью за счет активации гликогенолиза и глюконеоге неза; нарушение утилизации глюкозы в дихотомическом и апотомиче ском путях, реакциях гликогеногенеза и биосинтетических путях ли пидного обмена), увеличению уровня НЭЖК (активация липолиза в
303
Генетическая |
|
Факторы окружающей среды |
||
предрасположенность |
+ |
и образа современной жизни |
||
Ожирение, дислипопротеидемии, |
Гиподинамия; избыточное, |
|||
мутации генов K |
АТФ |
-каналов |
несбалансированное |
|
β-клеток, глюкокиназы, |
|
и нерегулярное питание; |
||
митохондриальной ДНК и др. |
|
хронический стресс и др. |
||
Инсулинорезистентность
Повышение продукции инсулина Избыточная активация β-клеток
Снижение числа β-клеток, нарушение толерантности к глюкозе, гипергликемия, активация мобилизации липидов, начальные проявления глюкотоксичности
Значительное уменьшение числа β-клеток, выраженные проявления глюкотоксичности с нарастанием гипергликемии и развитием осложнений
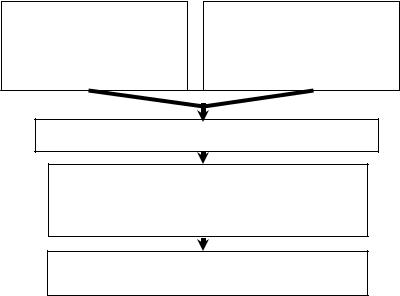
Рис. 35. Патогенез сахарного диабета 2-го типа
жировой ткани). Указанные изменения во многом реализуются через повышение продукции контринсулярных гормонов, и их можно рас сматривать как защитно-приспособительные. Вследствие нарушения утилизации глюкозы β-клетками продукция инсулина повышается. В конечном итоге развиваются нарушения функций β-клеток (вначале с отсутствием изменения их количества, а в последующем с его сниже нием). Итак, основными особенностями сахарного диабета 2-го типа являются инсулинорезистентность, гиперинсулинемия, дисфункция β клеток, в то время как уровни инсулина и гликемический контроль варьируют в зависимости от стадии заболевания и способности гипе ринсулинемии компенсировать высокий уровень глюкозы.
Заболевание чаще развивается после достижения 40-летнего возраста, однако в последние годы отмечается увеличение частоты развития сахарного диабета 2-го типа в более молодом возрасте и даже у подростков вследствие повышения частоты ожирения. Сахарный диабет 2-го типа тесно связан с другими метаболическими наруше ниями, в том числе с ожирением, артериальной гипертензией, дисли попротеидемиями, при этом заболевании более быстро и тяжело раз
304
вивается атеросклероз с его грозными осложнениями в виде инфаркта миокарда и инсультов.
Экспериментальные модели сахарного диабета 2-го типа
уживотных
Кнастоящему времени создано относительно большое коли чество экспериментальных моделей спонтанно развивающегося и ге нетически обусловленного сахарного диабета 2-го типа у животных. В большинстве из этих моделей комбинируются два главных проявления этого типа диабета: 1) инсулинорезистентность, ассоциированная с ожирением, 2) нарушение функций β-клеток в сочетании с уменьше ние общего количества β-клеток или без него. Эти модели имеют, с одной стороны, много общих закономерностей с проявлением сахар ного диабета 2-го типа у человека, а с другой – ряд отличий. Тем не
менее, они удобны для выяснения патогенеза этого заболевания у че ловека и создания новых лекарственных препаратов.
Важная особенность многих моделей сахарного диабета 2-го типа у животных – наличие выраженного полового диморфизма. У самцов в сравнении с самками во многих экспериментальных моделях развивается значительно более тяжелое заболевание. Для людей, на оборот, характерно преобладание и более тяжелое течение сахарного диабета 2-го типа у женщин в сравнении с мужчинами.
Для разработки новых экспериментальных моделей этой фор мы диабета важную роль сыграли подходы с повреждением (нокау том) определенных генов, а также с их переносом (трансгенные жи вотные).
1. Модели спонтанного сахарного диабета 2-го типа
Мутации гена лептина (мыши Lepob) и рецептора к лептину
(мыши db). Мутация гена лептина Lepob (ранее ее обозначали как ob) впервые описана в 1950 г., но идентификация гена проведена значи тельно позднее, после открытия лептина. У мышей, гомозиготных по этой мутации, лептин не синтезируется. Для них характерно развитие выраженной гиперфагии (повышение потребления пищи), ожирения, инсулинорезистентности и гиперинсулинемии. У животных развива ются множественные нарушения функций гипоталамуса, приводящие как к расстройствам метаболизма и ожирению, так и к бесплодию. Введение лептина приводит к снижению потребления пищи и к вос становлению многих нарушений обмена веществ. Инсулинорезистент ность выявляется при исследовании влияния инсулина на метаболизм мышечной и жировой ткани, печени, а метаболические изменения в
305
них отменяются при введении животным инсулина. Гипергликемия и выраженная гиперинсулинемия сочетается с развитием гиперплазии β клеток, общее количество которых повышается в 10 раз.
У мышей db выявлены множественные дефекты рецептора к лептину. Как и у мышей Lepob, у db-мышей развиваются гиперфагия и ожирение, гиперинсулинемия, а в возрасте 6–8 недель – гиперглике мия вследствие снижения функции β-клеток. Заболевание протекает более тяжело, чем у мышей Lepob. Терапия синтетическими лекарст венными препаратами, снижающими инсулинорезистентность (в част ности, производными тиазолидиндионов), предупреждает развитие диабета.
Мыши с мутацией гена агути. У мышей с доминантной «жел той» мутацией гена агути, определяющего характерную для этого вида животных серую (или агути) окраску шерсти и кожи, развивается ожи рение и гипергликемия. В зависимости от исходной линии мышей му тация гена агути приводит к варьирующим изменениям фенотипа. У мышей с повышенной предрасположенностью гиперинсулинемия раз вивается в возрасте 6 недель, в дальнейшем уровень инсулина про должает нарастать, при гистологическом исследовании отмечается β клеточная гиперплазия и гипертрофия. Характерна системная продук ция белков, в норме выявляющихся только в коже, что связано со вставкой в промоторный регион гена агути. Несколько групп генов, включая гены ферментов синтеза жирных кислот, имеют в промотор ном регионе отвечающие элементы и для инсулина и для окраски агу ти. Поэтому характерно значительное увеличение синтеза жирных кислот в печени и их повышенное использование для синтеза жира в жировых клетках. Функция гена агути к настоящему времени оконча тельно не выяснена, но у животных с его мутацией отмечаются гипер фагия и ускоренный рост.
Мыши KK. Были первоначально выведены вследствие не обычно больших размеров, но у большинства из них ожирение выра жено не в большей степени, чем у других мышей obese (масса чаще меньше 60 г). При скрещивании KK-мышей с животными разных ис ходных линий у потомков выраженность резистентности к инсулину, гиперинсулинемии и гипергликемии варьирует. Наиболее исследована линия мышей KKAy, полученная в Японии. У мышей этой линии зна чительно повышается концентрация инсулина в крови (более 1000 мкЕД/мл). Мутация, ответственная за KK-фенотип, не изучена.
Новозеландские тучные мыши (NZO, англ. New Zealand obese mice). Получены методом инбридинга (близкородственного скрещива ния) тучных аутбредных мышей. Новорожденные NZO-мыши имеют
306
повышенную массу тела. Животные обоего пола более крупные, чем обычные мыши, и имеют повышенное количество жировой ткани. При скрещивании мышей без дефекта с NZO-самцами (но не с самками этой линии) у 40–50% потомков развивается сахарный диабет 2-го типа в возрасте 12–20 недель при их кормлении диетой, содержащей 4,5% жира. Ожирение у мышей линии NZO характеризуется выражен ным увеличением подкожной и висцеральной жировой ткани. Инсули норезистентность в отличие от инсулинорезистентности у мышей Lep tob и db характеризуется неизмененным ответом на инсулин генов, кодирующих большинство ключевых ферментов глюконеогенеза и гликолиза в печени, однако выявляется нарушение активности фрукто зо-1,6-бисфосфатазы. Описано нарушение секреции инсулина β клетками как in vivo, так и in vitro, связанное с нарушением гликолиза, что ведет к дефекту высвобождения инсулина под воздействием глю козы. Генетические нарушения у NZO-мышей являются полигенными. Характерно развитие аутоиммунных нарушений, в том числе повреж дение почек, сходное с системной красной волчанкой, а также появле ние аутоантител против инсулинового рецептора. Показано, что от дельные проявления заболевания развиваются и у мышат, вскармли ваемых грудным молоком мышей NZO.
Рассмотренные модели спонтанного сахарного диабета 2-го типа у мышей являются наиболее известными, их полный список от нюдь не исчерпывается вышеприведенными примерами. Работа в этом направлении постоянно продолжается. Кроме животных со спонтан ным сахарным диабетом 2-го типа, сопровождающимся ожирением, выведены линии мышей с развитием заболевания без ожирения.
Помимо экспериментальных моделей у мышей создано не сколько линий крыс, мини-свиней, обезьян со спонтанным сахарным диабетом 2-го типа. Заболевание у этих животных также в зависимо сти от линии протекает либо с ожирением, либо без него.
2. Модели сахарного диабета 2-го типа на генетически модифицированных животных (нокаут генов / трансгенные животные)
Показано развитие сахарного диабета 2-го типа в сочетании с ожирением у мышей с нокаутом гена β3-адренорецепторов. Для моде лирования сахарного диабета 2-го типа без ожирения использованы трансгенные и генетически нокаутированные животные (главным об разом мыши) с модификацией генов, ответственных:
1) за развитие инсулинорезистентности, в частности генов, кодирующих IRS-1, IRS-2, которые участвуют в передаче сигнала с инсулинового рецептора, а также гена транспортера глюкозы в жиро-
307
вой и мышечной тканях GLUT-4;
2)за метаболизм липидов и глюкозы, в частности генов ядер ных рецепторов PPARs (англ. peroxisome-proliferator-activated receptors);
3)за регуляцию секреции инсулина, в частности генов транс портера глюкозы в β-клетках поджелудочной железы GLUT-2, глюко киназы, рецептора инсулиноподобного фактора роста-1 (IGF-1R).
Впоследние годы интенсивно исследуется сахарный диабет и
утрансгенных грызунов с геном человеческого островкового амило идного полипептида (англ. human islet amyloid polypeptide, hIAPP).
3. Модели индуцированного сахарного диабета 2-го типа
Диабет, индуцированный у мышей введением ауротиоглюко-
зы. Является примером химически индуцированного сахарного диабе та. Ауротиоглюкоза (AuSC6H11O5) – лекарственный препарат, перво начально предложенный для лечения ревматоидного артрита. Введе ние ауротиоглюкозы мышам приводит к повреждению вентромеди ального гипоталамуса с последующим развитием гиперинсулинемии, гиперфагии, ожирения, инсулинорезистентности. Уровень глюкозы вначале снижается, а затем повышается. Эта экспериментальная мо дель может служить примером регуляторной панкреатической дис функции, приводящей к формированию инсулинорезистентности с последующей ее компенсацией.
Сахарный диабет 2-го типа, индуцированный хирургическими вмешательствами. После хирургического повреждения вентромеди ального гипоталамуса на фоне кормления крыс специальной диетой, обогащенной липидами, у них развивается сахарный диабет 2-го типа с ожирением. Другой моделью является сахарный диабет, развиваю щийся у животных после операции частичной панкреатэктомии.
Сахарный диабет 2-го типа, развивающийся у крыс при введении стрептозотоцина в субпороговой дозе (40 мг/кг однократно) и замене питьевой воды на 10% раствор фруктозы. Учитывая, что ме ждународный обмен животными со спонтанным диабетом (в том чис ле, приобретение за рубежом племенного ядра с последующей транс портировкой по международным правилам пограничного ветеринар ного контроля) – процедура крайне сложная в организационном пла не, эта модель, хорошо воспроизводимая на обычных белых лабора торных крысах, представляет значительный интерес. Авторы из Юж но-Африканской Республики D. Rachel, R.D. Wilson, M.S. Islam пред ставили, на наш взгляд, убедительные доказательства развития при введении крысам стрептозотоцина в субпороговой дозе (40 мг/кг мас
308

сы тела однократно внутрибрюшинно) в сочетании с заменой питьевой воды на 10% раствор фруктозы сахарного диабета 2-го типа, а также возможности использования этой экспериментальной модели для оценки биологического действия новых противодиабетических лекар ственных препаратов2. Развитие инсулинорезистентности при ограни ченном химическом повреждении β-клеток поджелудочной железы на фоне воздействия фруктозы связано с тем, что концентрация этого моносахарида в крови контролируется через другие нейроэндокрин ные механизмы, чем уровень глюкозы. В плане выяснения роли раз личных факторов в развитии диабета 2-го типа представляются важ ными данные о диабетогенном действии фруктозы, так как этот угле вод входит в состав многих пищевых продуктов и безопасность его в высоких концентрациях ранее не изучалась. Более того, фруктоза в настоящее время используется как заменитель сахара у больных са харным диабетом.
Общие механизмы нарушения метаболизма при сахарном диабете
Независимо от того, является ли недостаточность инсулина абсолютной (снижение концентрации инсулина в циркуляции и в тка нях вследствие уменьшения его секреции поджелудочной железой) или относительной (инсулинорезистентность из-за нарушения переда чи сигнала с мембранного рецептора к инсулину внутрь клетки), са харному диабету присущи общие механизмы нарушения обмена ве ществ, связанные с одинаковыми изменениями в конечных звеньях регуляторной цепи на внутриклеточном уровне.
Для сахарного диабета характерны нарушения всех видов об мена веществ: углеводного, липидного, аминокислотного и белкового, водно-электролитного.
Типовые нарушения углеводного обмена. Дефицит инсулина или нарушение его действия на клетки-мишени приводят к нарушению утилизации тканями ключевого метаболита всего углеводного обмена
– глюкозы. В норме уровень глюкозы в крови натощак после сна со ставляет 3,33–5,55 ммоль/л. Глюкоза крови используется для энерге тических и пластических целей в основном следующими шестью ор ганами и тканями: мозгом (40–60%), скелетной мускулатурой и мио кардом (15–20%), почками (10–15%), клетками крови (5–10%), пече нью и органами брюшной полости (3–6%), жировой тканью (2–4%). В
2 Rachel D., Wilson R.D., Islam M.S. Fructose-fed streptozotocin-injected rat: an alternative model for type 2 diabetes // Pharmacological Reports. 2012. Vol. 64(1). P. 129–139.
309
транспорте глюкозы через клеточные мембраны ключевую роль игра ют мембранные транспортеры глюкозы: GLUT1, 2, 3, 4 и SGLT1, 2. Семейство транспортеров GLUT обеспечивает облегченную диффу зию глюкозы, не зависящую от энергии и подчиняющуюся кинетике Михаэлиса – Ментен. Высокоаффинные транспортеры (GLUT 1, 3, 4) имеют значение константы Михаэлиса – Ментен (Km) ниже нормаль ного уровня глюкозы и обеспечивают транспорт глюкозы во многих клетках в базальных условиях. GLUT3 – это главный нейральный транспортер (с наименьшей Km), тогда как GLUT4 обеспечивает ин сулинстимулированный транспорт глюкозы клетками скелетной мус кулатуры, сердца и жировой ткани. Инсулин и физическая нагрузка усиливают экспрессию GLUT3 на клеточной мембране. Низкоаффин ный транспортер (GLUT2) присутствует на ß-клетках поджелудочной железы, а также в органах с высокой скоростью транспорта глюкозы, таких как кишечник, печень и почки. Транспортеры семейства SGLT используют электрохимический натриевый градиент для транспорта глюкозы против градиента концентрации в основном в кишечнике и в почках. SGLT1 ответственен за транспорт глюкозы из тонкого кишеч ника, тогда как SGLT2 играет главную роль в реабсорбции глюкозы в проксимальных канальцах почек. При недостаточности инсулина транспорт глюкозы инсулинзависимыми транспортерами нарушается.
В мышцах и печени глюкоза используется для окисления в реакциях гликолиза и дихотомического пути, а в печени дополнитель но – в пентозном цикле, необходимом для образования НАДФН+Н+, выступающего в качестве источника восстановительной энергии для биосинтеза жирных кислот, холестерина, желчных кислот, витамина D3, других липидов. Избыток глюкозы в печени и мышцах использует ся для синтеза гликогена. То количество глюкозы, которое не было израсходовано для реакций катаболизма и синтеза гликогена, утилизи руется жировой тканью для синтеза триацилглицеролов. При повыше нии потребности организма в энергии, например при интенсивной фи зической нагрузке, гликоген расщепляется под действием контринсу лярных гормонов для глюкозы в печени и лактата в мышцах. При сни жении уровня глюкозы крови, а также под действием глюкокортикои дов при стрессе в печени происходит синтез глюкозы и гликогена из лактата, пирувата, кислот цикла Кребса, аминокислот в результате процессов глюконеогенеза. Инсулин усиливает реакции катаболизма глюкозы за счет активации ключевых ферментов гликолиза и гликоли тической фазы дихотомического пути (гесокиназы, фосфофруктокина зы, пируваткиназы), цикла Кребса (прежде всего – цитратсинтазы), пентозного цикла (глюкозо-6-фосфатдегидрогеназы и 6
310
фосфоглюконатдегидрогеназы). Активация инсулином этих же путей в жировой ткани способствует образованию центрального метаболита для синтеза липидов – ацетил-коэнзима А, а также обеспечивает син тез липидов энергетически за счет образования НАДФН+Н+. Инсулин ингибирует ключевые ферменты глюконеогенеза (пируваткарбоксила зу, фосфоенолпируваткарбоксикиназу, фруктозо-1,6-бисфосфатазу, глюкозо-6-фосфатазу), гликогенолиза (фосфорилазу), активирует клю чевой фермент синтеза гликогена – гликогенсинтетазу.
Нарушение утилизации глюкозы тканями и вышеперечислен ных метаболических путей при недостаточности инсулина, а также активация контринсулярными гормонами глюконеогенеза приводит к развитию гипергликемии, а при превышении почечного порога для глюкозы (9–10 ммоль/л) к появлению ее в моче (глюкозурия). Дли тельное повышение уровня глюкозы в крови и тканях при сахарном диабете приводит к усилению гликозилирования белков, что лежит в основе развития целого ряда осложнений. Важным показателем для оценки степени выраженности реакций гликозилирования белков яв ляется увеличение концентрации в крови гликозилированного гемо глобина (этот параметр в настоящее время пациенты с сахарным диа бетом вместе с концентрацией глюкозы и кетоновых тел могут опре делять сами с помощью некоторых моделей индивидуальных глюко метров). Помимо этого значительное повышение уровня глюкозы в крови может привести к развитию гиперосмолярной комы. Энергети ческая недостаточность клеток и тканей через глюкорецепторы приво дит к развитию полифагии (повышение потребления пищи) и булемии (чувство «волчьего» голода). Снижение активности пируватдегидроге назного комплекса при сахарном диабете ведет к нарушению декар боксилирования пирувата, увеличению концентрации пирувата и лак тата (пируватацидемии и гиперлактатацидемии), что при тяжелых формах диабета может привести к развитию лактатацидоза и лактата цидотической комы.
Типовые нарушения липидного обмена. Дефицит инсулина приводит к нарушению синтеза жирных кислот (ключевой фермент – ацетил-КоА-карбоксилаза), синтеза триацилглицеролов, синтеза холе стерина и его производных. Важную роль в нарушении биосинтетиче ских реакций жирового обмена играет снижение образования НАДФН+Н+ в пентозном цикле. Компенсаторное повышение уровня контринсулярных гормонов в ответ на энергетический голод ведет к усилению липолиза, мобилизации НЭЖК, β-окисления жирных кислот с образованием ацетил-КоА. Избыток последнего не может в условиях недостаточности инсулина утилизироваться в цикле Кребса и в био
311
синтетических реакциях липидного обмена. Поэтому через образова ние гидрооксиметилглутарил-КоА усиливается синтез кетоновых тел – ацетоацетата, β-гидроксибутирата и ацетона. Это приводит к развитию гиперкетонемии и гиперкетонемического ацидоза. Кетоновые тела появляются также в моче (кетонурия). При тяжелом течении сахарного диабета может развиться гиперкетонемическая кома.
При сахарном диабете 2-го типа развивается гиперхолестери немия, увеличение уровня НЭЖК приводит к повышению образования липопротеидов очень низкой плотности (ЛПОНП), богатых триацил гицеролами, что, в свою очередь, ведет к увеличению уровня послед них в крови. Триацилглицеролы переносятся на липопротеиды низкой плотности (ЛПНП) и на липопротеиды высокой плотности (ЛПВП). Повышается катаболизм ЛПВП и аполипопротеина А. Увеличение уровня ЛПНП, обогащенных триацилглицеролами, способствует раз витию атеросклероза (макроангиопатии).
Типовые нарушения аминокислотного и белкового обмена.
Дефицит инсулина ведет к нарушению синтеза заменимых аминокис лот из метаболитов углеводного обмена, а повышение вследствие энергетического голода тканей уровня контринсулярных гормонов – к увеличению катаболизма аминокислот и их использования для глюко неогенеза. Увеличивается катаболизм белков, в частности белков мы шечной ткани, выпадает анаболическое действие инсулина на биосин тез белков и нуклеиновых кислот. Вследствие увеличения катаболизма белков, аминокислот и липидов при сахарном диабете 1-го типа разви вается похудение.
Типовые нарушения водно-электролитного обмена. Развитие гиперосмолярности при сахарном диабете приводит к полидипсии (жажда), полиурии (повышение суточного диуреза), обезвоживанию. Развитие полиурии связано с увеличением осмотического диуреза, а также с изменением продукции гормонов, регулирующих водно электролитный обмен: альдостерона, антидиуретического гормона, ренина, ангиотензина. Развиваются гипернатриемия, в последующем – нарушение функции почек, снижение объема циркулирующей крови.
Критерии диагноза сахарного диабета
• Симпотомы сахарного диабета + концентрация глюкозы в капиллярной крови натощак превышает 7,0 ммоль/л, а через 2 ч после приёма пищи – 11,1 ммоль/л; в результате проведения глюкозотоле рантного теста (после приема 75 г глюкозы) уровень глюкозы крови превышает 11,1 ммоль/л (необходим повтор);
312
•уровень гликозилированного гемоглобина превышает 5,9% (5,9–6,5% – сомнительно, более 6,5% большая вероятность);
•глюкозурия.
Осложнения сахарного диабета
Острые осложнения:
•диабетический кетоцидоз и гиперкетонемическая кома;
•гипогликемия и гипогликемическая кома;
•некетонемический гиперосмолярный синдром (повышение осмотического давления вследствие гипергликемии в сочетании с обезвоживанием и нарушением водно-электролитного обмена, в тяже лых случаях развивается гиперосмолярная кома);
•лактацидотический ацидоз (в тяжелых случаях кома);
•острые инфекции.
Поздние осложнения:
•диабетические микроангиопатии – повреждение мелких со судов с нарушением их проницаемости, повышением их ломкости, склонностью к кровоизлияниям, образованию новых сосудов;
•диабетические макроангиопатии – повреждение более круп ных сосудов со склонностью к тромбозам, развитие атеросклероза и его осложнений;
•диабетическая ретинопатия – патология сетчатой оболочки глаза, частая причина слепоты у пациентов среднего и пожилого воз раста, которая характеризуется поражением сетчатки глаза в виде мик роаневризм, точечных и пятнистых кровоизлияний, твёрдых экссуда тов, отёка, образования новых сосудов; могут развиться кровоизлия ния на глазном дне и отслоение сетчатки; чаще встречается при сахар ном диабете 2-го типа, степень её выраженности коррелирует с выра женностью нефропатии; начальные стадии ретинопатии выявляются у 1/4 больных с впервые выявленным сахарным диабетом 2-го типа;
•диабетическая офтальмопатия – патология органа зрения, кроме ретинопатии, включает в себя раннее развитие катаракты (по мутнение хрусталика);
•болезнь Альцгеймера – заболевание центральной нервной системы, проявляется выраженной деменцией;
•диабетическая полинейропатия – поражение нервной систе мы, чаще всего проявляется в виде двусторонней периферической ней ропатии по типу «перчаток и чулок», начинающейся в нижних частях конечностей; потеря болевой и температурной чувствительности –
313
наиболее частая причина развития вывихов суставов и нейропатиче ских язв;
•диабетическая нефропатия – патология почек, проявляющая ся вначале в виде микроальбуминурии (появление альбумина в моче), затем протеинурии (наличие в моче белков с большей молекулярной массой, относящихся к глобулиновым фракциям); исходом может быть развитие хронической почечной недостаточности;
•диабетическая артропатия – патология суставов с появлени ем в них боли, «хруста», ограничения подвижности, уменьшения ко личества синовиальной жидкости и повышения её вязкости;
•инфекционные поражения кожи – вызываются условно патогенной микрофлорой, проявляются в виде рожистого воспаления, фурункулов, карбункулов, инфекций, вызванных грибами;
•инфекционные поражения слизистых оболочек – вызываются условно-патогенной микрофлорой, проявляются в виде молочницы, бронхитов, циститов и других инфекций на территории слизистых; сахарный диабет также характеризуется снижением резистентности ко многим инфекционным заболеваниям;
•диабетическая стопа – тяжелое поражение стоп больного сахарным диабетом в виде гнойно-некротических процессов, язв, ко стно-суставных поражений, возникающее на фоне изменения перифе рических нервов, сосудов, кожи и мягких тканей, костей и суставов, вторичной иммунной недостаточности; является основной причиной ампутаций у пациентов.
Иммунная система и сахарный диабет
Рассматривая участие иммунной системы в патогенезе сахар ного диабета, необходимо выделить два основных аспекта:
1)участие иммунной системы в патогенезе сахарного диабета как 1-го, так и 2-го типа;
2)формирование вторичной иммунной недостаточности при сахарном диабете.
Первому аспекту в настоящее время в мировой литературе уделяют основное внимание. Как указывалось выше, аутоиммунное повреждение β-клеток играет главную роль в патогенезе типа 1A (им муноопосредованного) сахарного диабета 1-го типа. Аутоиммунные реакции участвуют в патогенезе и других форм сахарного диабета как 1-го, так и 2-го типа, вызванного другими этиологическими факторами (вирусная инфекция, химические факторы и пр.), подключаясь к по вреждению β-клеток на более поздних стадиях развития заболевания. Наряду с аутоиммунной агрессией, важную роль в формировании ин
314
сулинорезистентности и нарушении утилизации глюкозы тканями на фоне гиперинсулинемии при диабете 2-го типа могут играть провоспа лительные цитокины (TNF-α, IL-1β, IL-6).
Второму аспекту, несмотря на его важность, уделяется мень шее внимание. Хотя развитие вторичной иммунной недостаточности при сахарном диабете как 1-го, так 2-го типа не вызывает сомнения, а ее клиническим проявлением является значительное снижение рези стентности организма к инфекциям, вызванным вирусами, грамполо жительными и грамотрицательными бактериями, микобактериями ту беркулеза, анаэробными бактериями, грибами и другими, в том числе условно-патогенными, микроорганизмами, механизмы развития им мунодефицитного состояния изучены недостаточно. Также требуют дальнейшей разработки и подходы к иммунокорригирующей терапии при инфекционных осложнениях. Хотя в работах, опубликованных еще в 80–90-е гг. XX в., показано развитие при сахарном диабете на рушения функций Т-лимфоцитов и фагоцитирующих клеток, а также продемонстрирована способность инсулина активировать тимусзави симый иммунный ответ у мышей, в том числе в условиях неизменен ного уровня глюкозы (Н. Н. Кеворков с соавт., 1980; Н. Н. Кеворков, 1995), роль отдельных звеньев нейроэндокринной регуляции функций клеток иммунной системы в развитии инфекционных осложнений при сахарном диабете изучена недостаточно.
В наших исследованиях, выполненных в 2012 г. при поддерж ке фонда «Нева» в рамках программы сотрудничества по вопросам метаболизма и диабета с Федеральной политехнической школой Ло занны (Швейцария), установлено, что при сахарном диабете 1-го типа, индуцированном у самцов белых нелинейных крыс однократным вну трибрюшинным введением стрептозотоцина в дозе 65 мг/кг, на 10-е сутки эксперимента развивается резко выраженное снижение проли феративного ответа лимфоцитов селезенки в культурах с конканава лином А (Т-клеточный митоген), митогеном лаконоса (pokeweed mitogen, тимусзависимый В-клеточный митоген, активирующий поли клональную пролиферацию и дифференцировку В-лимфоцитов в плазматические клетки при участии Т-хелперов) и липополисахаридом Salmonella enterica серотипа typhimurium (тимуснезависимый В клеточный митоген) (Шилов Ю.И. с соавт., 2013). В исследованиях С.В. Гейна с соавт., выполненных в рамках этого же проекта, установ лено, что при стрептозотоциновом диабете у мышей наряду со сниже нием пролиферативного ответа спленоцитов в культурах с конканава лином А развивается выраженное снижение продукции интерферона гамма при отсутствии статистически значимого изменения синтеза
315
IL-4, IL-17 и IL-1β в 48-часовых культурах спленоцитов с тем же ми тогеном. Сходные по направленности изменения функций иммунной системы выявлены и в исследованиях Б. А. Бахметьева с сотр. (2013) при гестационном диабете у женщин, выполненных в 2012 г. по дру гому проекту при поддержке фонда «Нева» в рамках той же програм мы. Однотипность направленности изменения функций иммунной системы при разных формах сахарного диабета у экспериментальных животных и людей, на наш взгляд, не является случайной. Несмотря на различие патогенеза (диабет 1-го и 2-го типов, гестационный диабет и диабет других типов), на уровне клеток-мишеней результирующие метаболические нарушения носят однотипный xapaктep, обусловлен ный сходством изменений внутриклеточных сигнальных путей и экс прессией генов, контролируемой этими путями. С нашей точки зрения, именно это может определять типовые нарушения иммунитета при разных формах сахарного диабета. Дальнейшие исследования меха низма выявленных нарушений перспективны для разработки новых подходов к коррекции изменения функций иммунной системы и тера пии инфекционных осложнений при этом заболевании.
316