21. Циклічні нуклеотиди (цАмф, цГмф) як регулятори ферментативних реакцій та біологічних функцій клітини
В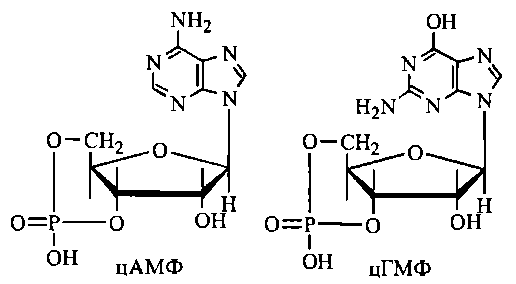 ажливою
та поширеною біологічною системою
контролю за ферментативними реакціями,
що поєднує в собі різні молекулярні
механізми регуляції, є система циклічних
нуклеотидів. Циклічні нуклеотиди
3',5'-АМФ (цАМФ) та 3',5' ГМФ (цГМФ) - це
внутрішні (3'5' дифосфорні ефіри аденілової
(АМФ) та гуанілової (ГМФ) кислот. Найбільш
поширеними є цАМФ-залежні системи
контролю за внутрішньоклітинними
біохімічними процесами, зокрема за
такими, що підлягають нейрогуморальній
регуляції з боку цілісного організму,
яка реалізується гормонами та
нейромедіаторами. Регуляція ферментативних
процесів за участю цАМФ включає декілька
послідовних
ажливою
та поширеною біологічною системою
контролю за ферментативними реакціями,
що поєднує в собі різні молекулярні
механізми регуляції, є система циклічних
нуклеотидів. Циклічні нуклеотиди
3',5'-АМФ (цАМФ) та 3',5' ГМФ (цГМФ) - це
внутрішні (3'5' дифосфорні ефіри аденілової
(АМФ) та гуанілової (ГМФ) кислот. Найбільш
поширеними є цАМФ-залежні системи
контролю за внутрішньоклітинними
біохімічними процесами, зокрема за
такими, що підлягають нейрогуморальній
регуляції з боку цілісного організму,
яка реалізується гормонами та
нейромедіаторами. Регуляція ферментативних
процесів за участю цАМФ включає декілька
послідовних
стадій передавання і трансформації хімічного (регуляторного) сигналу.
1. Утворення циклічних нуклеотидів у реакціях, що каталізуються ферментами циклазами: аденілатциклазою та гуанілатциклазою з нуклеозидтрифосфатів АТФ та ГТФ, відповідно:
Р![]() озщеплення
цАМФ та цГМФ до звичайних, нециклічних
нуклеозидмонофосфатів каталізується
фосфодіестеразою циклічних нуклеотидів.
Фермент аденілатциклаза розміщений у
плазматичних мембранах клітин і його
активація відбувається в результаті
взаємодії з рецепторами мембран певних
фізіологічно активних сполук, зокрема
гормонів адреналіну, глюкагону тощо.
озщеплення
цАМФ та цГМФ до звичайних, нециклічних
нуклеозидмонофосфатів каталізується
фосфодіестеразою циклічних нуклеотидів.
Фермент аденілатциклаза розміщений у
плазматичних мембранах клітин і його
активація відбувається в результаті
взаємодії з рецепторами мембран певних
фізіологічно активних сполук, зокрема
гормонів адреналіну, глюкагону тощо.
2. Активація циклічним АМФ протеїнкіназ, функцією яких є фосфорилування інших ферментних білків. Ці цАМФ-залежні протеїнкінази є регуляторними ферментами, що активуються цАМФ за механізмом алостеричного контролю.
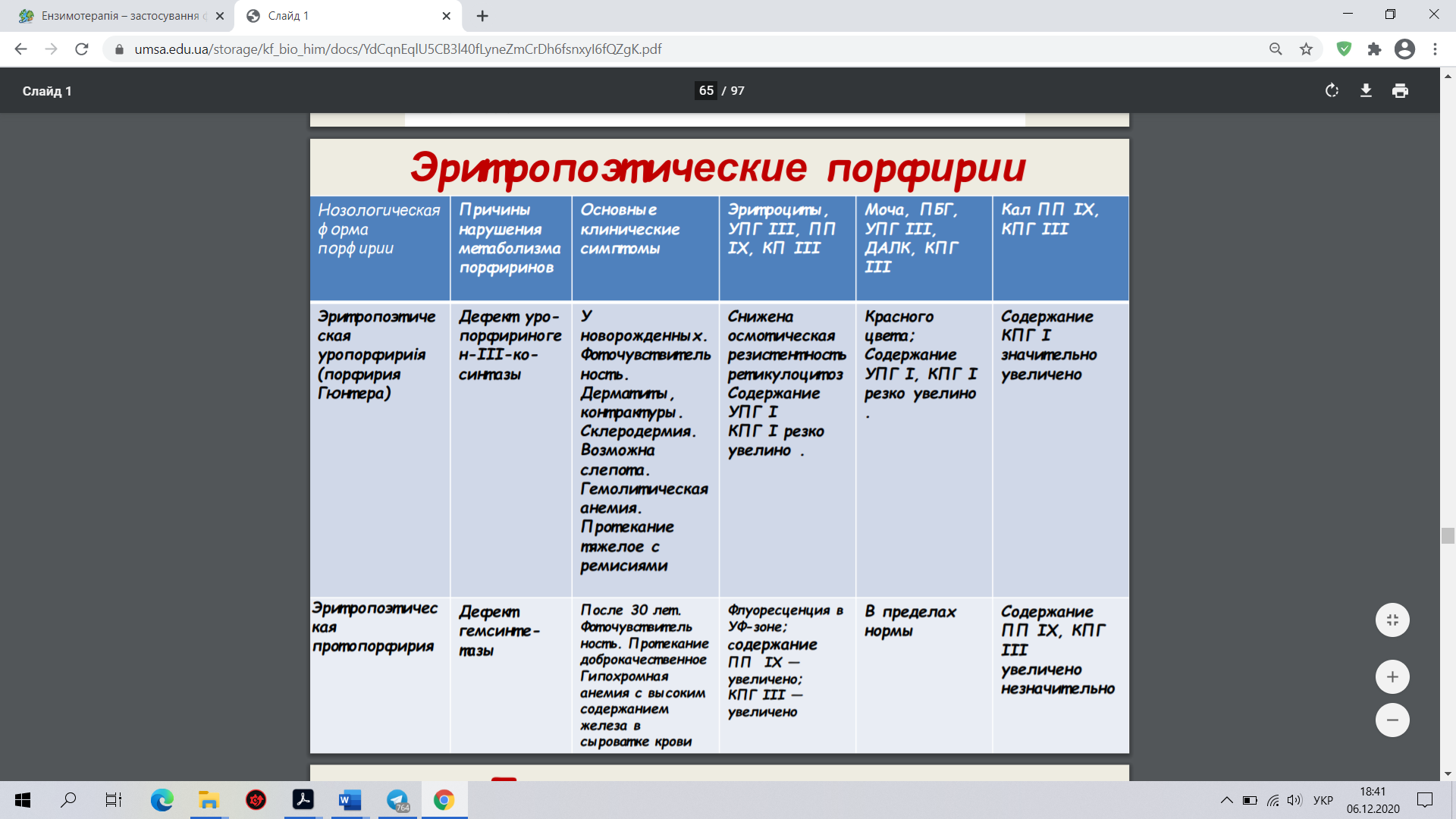
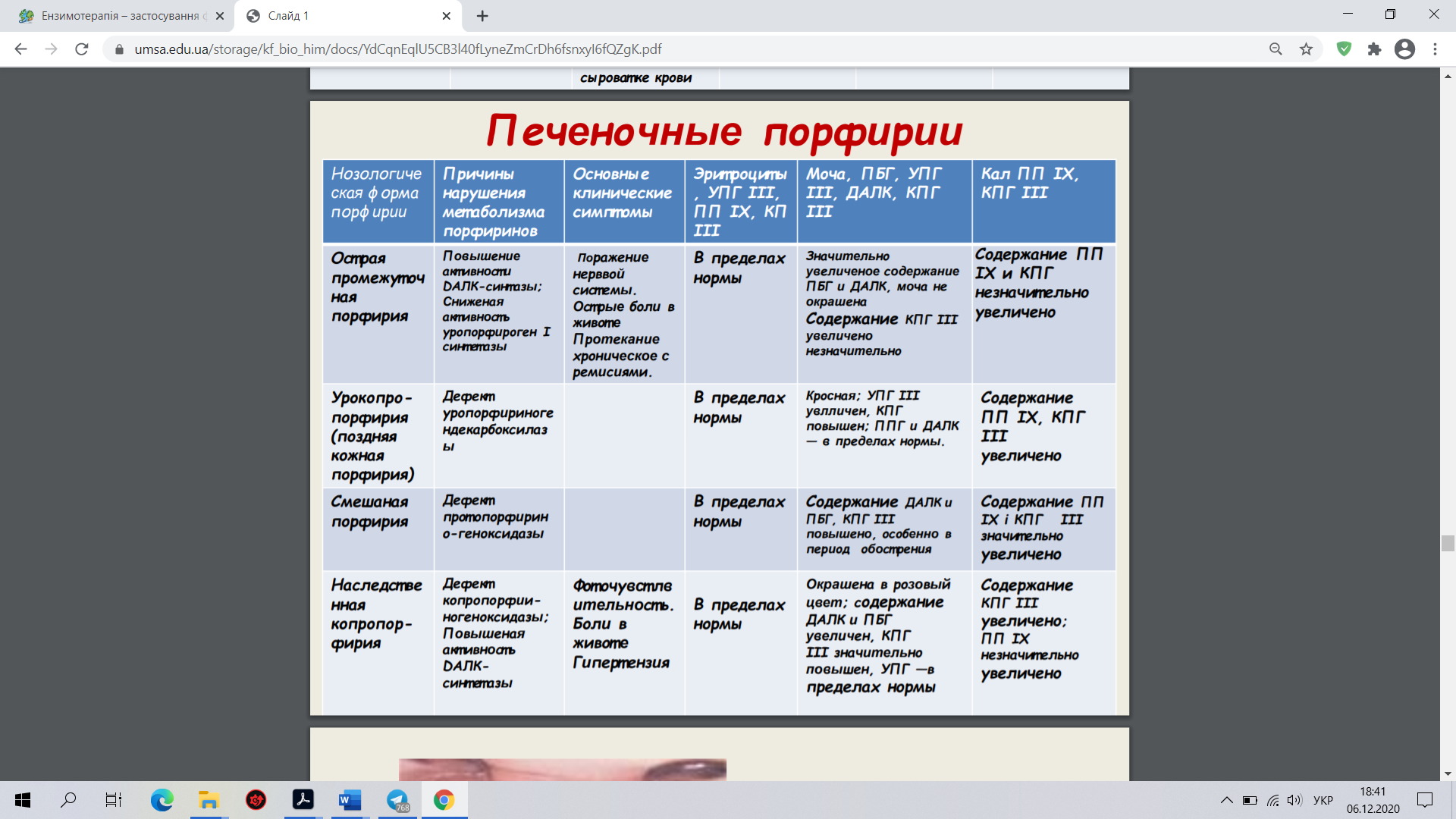
22. Ензимопатії – уроджені (спадкові) вади метаболізму вуглеводів, амінокислот, порфіринів, пуринів
Первинні, або спадкові, ензимопатії виникають унаслідок змін у генетичному коді синтезу ферментів. Причинами ферментативних дефектів можуть бути: аномальна структура ДНК, порушення перенесення генетичного коду від ДНК до РНК, змінена структура РНК і порушення в передачі інформації від РНК до рибосом. Крім того, причиною метаболічних розладів можуть бути генетично зумовлені порушення співвідношення природних активаторів та інгібіторів ферментів. Причиною спадкових ензимопатій є мутації, що виявляються характерними змінами в активності відповідних ферментів. При цьому ферментативна активність відсутня або знижена, або (дуже рідко) підвищена. Можуть з’являтися патологічні ферменти, які в нормі не трапляються.
1. Галактоземії (дефіцит галактозо- 1-фосфатуридилтрансферази, або галактокінази). При цій патології відбувається накопичення в крові й тканинах галактозо-1-фосфату, вільної галактози та спирту дульциту продукту відновлення галактози. Високий їх уміст діє токсично, у немовлят після споживання молока спостерігають блювання й пронос, збільшується печінка, розвивається катаракта, затримується розумовий розвиток.
2. Фруктоземії (дефіцит фруктозодифосфатальдолази, або фруктокінази). Генетичний дефект альдолази фруктозо-1-фосфату зумовлює істотні порушення в обміні вуглеводів, гіпоглікемію, ураження печінки.
3. Глікогенози (захворювання, зумовлені метаболічними порушеннями, які призводять до надмірної концентрації глікогену або зміни його структури):
I тип, гепаторенальний глікогеноз, хвороба Гірке (дефіцит глюкозо-6-фосфатази).
II тип, генералізований глікогеноз, хвороба Помпе (дефіцит кислої 1,4-а-глюкозидази). Спостерігають збільшення розмірів серця (кардіомегалія) з гіпотонією та серцево-легеневою недостатністю. Смерть настає в дитячому віці.
III тип, нирково-м’язовий глікогеноз, хвороба Корі (дефіцит аміло-1,6-глюкозидази).
IVтип, печінково-циротичний ендотеліальний глікогеноз, хвороба Андерсена (дефіцит 1,4-а-глюкан-6-а-глюкозилтрансферази). У хворих спостерігають гепатомегалію та спленомегалію, печінкову недостатність.
V тип, м’язовий глікогеноз, хвороба Мак-Ардля (дефіцит м’язової фосфорилази).
4. Непереносність дисахаридів
Аглікогеноз (дефіцит глікогенсинтетази) характеризується гіпоглікемією з судомами, блюванням, порушенням розумового розвитку.
5.Мукополісахаридози, (група метаболічних захворювань сполучної тканини, пов'язаних з порушенням обміну кислих глікозаміногліканів, пов'язаних недостатністю лізосомних ферментів обміну глікозаміногліканів) різні типи (дефіцит глюкуронозилдисульфоглюкозамінглюкуронідази, сульфатази, N-ацетилгексозамінідази, ідуронідази, N-ацетилглюкозаміні дази).
6. Гемолітичні анемії, зумовлені дефіцитом ферментів обміну вуглеводів в еритроцитах.
1. Фенілкетонурія (дефіцит фенілаланін-4-монооксигенази). Блокада перетворення фенілаланіну в тирозин, накопичення фенілпірувату (токсична речовина для мозку) – відставання в розумовому розвитку.
2. Алкаптонурія (дефіцит гомогентизинат-1,2-діоксигенази). Це спадкове захворювання розвивається внаслідок генетичного дефекту гомогентизинат-1,2-діоксигенази — ферменту катаболізму фенілаланіну.
Фенилаланин → Тирозин ⇋ п-гидроксифенилпировиноградная кислота → Гомогентизиновая кислота → Малеилацетоуксусная кислота → Фумарилацетоуксусная кислота → Фумаровая кислота + Ацетоуксусная кислота
3. Тирозінемії
Рідше трапляються такі спадкові порушення, як тирозинемії, причиною яких являється дефіцит, або відсутність ферментів катаболічних шляхів перетворення тирозину в фумарат та ацетоацетат.
Тирозинемія І типу (дефіцит фумарилацетатгідроксилази) призводить до накопичення токсичних метаболітів, які знижують активність ряду ферментів та транспортних систем. Розвивається печінкова недостатність і нефропатія.
Тирозинемія ІІ типу або синдром Ріхтера-Ханхарта (дефіцит тирозинамінотрансферази) проявляється аномаліями рогівки ока та шкіри, затримкою розумового розвитку; клінічні симптоми визначаються накопиченням тирозину.
4.Альбінізм (дефіцит тирозинази) — молекулярна хвороба. Спадкова відсутність тирозинази призводить до альбінізму.
Тирозиназа катализирует синтез меланина и других пигментов из их предшественника тирозина.
5.Гіперамоніємія (дефіцит ферментів синтезу сечовини: карбамоїлфосфатсинтази, аргініносукцинатліази, орнітинкарбамоїлтрансферази, аргініносукцинатсинтетази, аргінази).
Захворювання |
Дефект ферменту |
Клінічні прояви |
Метаболіти |
|
Кров |
Сеча |
|||
Гіперамоніємія, І тип |
Карбамоїлфосфат-синтетаза |
Впродовж 24 - 48 годин після народження кома, смерть |
Глн, Ала, NН3 |
Оротат |
Гіперамоніємія, ІІ тип |
Орнітинкарбамоїл-трансфераза |
Гіпотонія, зниження толерантності до білків |
Глн, Ала, NН3 |
Оротат |
Цитрулінемія |
Аргініносукцинат-синтетаза |
Гіперамоніємія у новонароджених, у дорослих – після вживання білків |
Цитрулін NН3 |
Цитрулін |
Аргініносукци-натурія |
Аргініносукцинат-ліаза |
Гіперамоніємія, атаксія, судоми, випадання волосся |
Аргіно-сукцинат NН3 |
Аргіносукцинат, Глн, Ала, Ліз |
Гіпераргінінемія |
Аргіназа |
Гіпераргінінемія |
Арг, NН3 |
Арг, Ліз,орнітин
|
6. Гіпергістидинемія (дефіцит гістидинази). Це спадкове захворювання виникає внаслідок відсутності гістидинази, що каталізує окисне дезамінування гістидину.