

Р
Пери центрическая инверсия
Нормальная хромосома 6
ис. 8.13. Перицентрическая и парацентрическая инверсии в хромосоме 6.
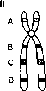
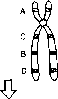
Е
СЖ
D
X
С В А я П И Ю
и м ■ ■
(
ВТ
с в и n -g
Е D
са:
в А
с
X
„О
ВТ.’" го
сж
в
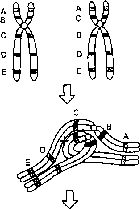
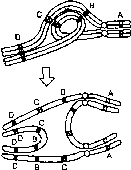
Рис. 8.14. Механизм образования рекомбинантных несбалансированных хромосом в случае перицентрической (I) и парацентрической (II) инверсии, когда кроссинговер происходит в инверсионной петле (из: R.F. Mueller, I.D. Young. Emery’s Elements of Medical Genetics. — Churchill Livingstone, 1998. — P. 50).
парацентрической инверсии, как видно из рис. 8.14, приведет к образованию либо ацентрического фрагмента хромосомы, либо дицентрика. Все 4 типа несбалансированных гамет в случае их участия в оплодотворении дадут нежизнеспособные зиготы, которые элиминируются на ранних стадиях развития. Вероятно, однако, что кроссинговер внутри инверсий может происходить крайне редко, так как конъюгация гомологичных хромосом при наличии в одной из них инверсии затруднена. У экспериментальных животных инверсии используют как раз для запирания кроссинговера.
Изохромосомы. Изохромосомы возникают в тех случаях, когда центромера делится не продольно, а поперечно. В результате одно из плеч теряется, а второе удваивается. Чаще всего выявляется изохромосома, составленная из длинных плеч хромосомы X. В этом случае у индивидуума, носителя такой изохромосомы X, обнаруживают проявления синдрома Шере- шевского—Тернера.
Кольцевые хромосомы. Этот тип хромосомной мутации возникает в том случае, когда разрывы наблюдаются в обоих плечах какой-то хромосомы. Ацентрические фрагменты при этом теряются, а центральная часть хромосомы замыкается в кольцо. Если такая кольцевая хромосома образуется из аутосомы, то из-за отсутствия значительной доли генетического материала этой хромосомы гамета и зигота оказываются несбалансированными, что должно привести к ранней потере зародыша с кольцевой хромосомой. Если все-таки зародыш образуется, то кольцевая хромосома имеет тенденцию теряться во время митотических делений клеток. Как следствие, возникает мозаи- цизм по наличию в клетках кольцевой хромосомы.
Номенклатура хромосомных мутаций
Ранее мы уже указывали на то, что для описания кариоти- па существует система принятых сокращений. Это относится и к описанию хромосомных мутаций. В табл. 8.2 приведены некоторые принятые символы.
8.6. Хромосомные болезни
После того как в 1956 г. были разработаны методы анализа хромосом человека, за короткое время была установлена хромосомная природа целого ряда заболеваний, в том числе синдрома Дауна (47,XX/XY+21), синдрома Клайнфелтера (47,ХХУ), синдрома Шерешевского—Тернера (45,X) и некоторых других синдромов аутосомных трисомий. В настоящее время различают почти 1000 хромосомных синдромов. Их вклад в спонтанные аборты, неонатальную смертность и за-
Тип мутации
Д
Символ
del
dup
fra
inv
rob
елеция, например 46,XX,de\(5p) — женщина с делецией короткого плеча хромосомы 5Дупликация, например 46,XY,dup(ll)(ql2) — мужской кариотип с дупликацией сегмента ql2 хромосомы 11
Ломкий сайт
Изохромосома, например 46,X,i(Xq) — женский кариотип, одна из хромосом X представлена изохромосомой по длинному плечу
Инверсия, например 46,XY,inv(10)(pl3ql2) — мужской кариотип, перицентрическая инверсия с точками разрыва р13 и ql2
Робертсоновская транслокация, например, 45,XX,rob (14q21q) — женщина со сбалансированной робертсоновской транслокацией длинных плеч хромосом 14 и 27, или 4о,ХХ,- 14,+rob( 14q21q) — женщина с несбалансированной робертсоновской транслокацией, хромосома 14 не идентифицируется в кариотипе, + хромосома с транслокацией
К
r
t
Мозаицизм
Аутосомная
трисомия
ольцевая хромосома, например 46,XX, г(16) — женщина с кольцевой хромосомой 16Транслокация, например 46,XX, t(2;4)(q21;q21) — женщина с реципрокной трансплантацией, включающей длинное плечо хромосомы 2, начиная с сегмента 2q21, и длинное плечо хромосомы 4, начиная с сегмента 4q21
Например, 46,ХХ/47,ХХ,+21 — женщина, часть клеток у которой содержит нормальный кариотип, а другая часть — трисомию по хромосоме 21
Например, 47,XY,+13 — мужчина, кариотип которого содержит 47 хромосом, дополнительная хромосома — 13
болеваемость весьма значительный. По-видимому, не менее 50 % всех спонтанных абортов обусловлены хромосомными мутациями: частота хромосомных аномалий среди новорожденных составляет 0,8 %, а среди мертворожденных — 5 %.
Патогенез хромосомных болезней чрезвычайно сложен, поскольку он зависит от нарушений проявления большого числа генов, вовлеченных в любой тип цитогенетически различимых хромосомных мутаций. Есть, однако, еще одна особенность патогенеза хромосомных болезней, которая отличает их от моногенных заболеваний. Эта особенность обусловлена тем, что при хромосомных болезнях их симптомы, обычно проявляющиеся врожденными пороками развития, являются следствием так называемого эффекта дозы гена. Хотя в некоторых случаях возникновение хромосомной мутации может приводить к нарушению структуры генов, попадающих в точки разрывов,
доминируют либо триплоидия, либо гаплоидия по большему или меньшему числу нормальных генов. В общем очевидно, что избыток генетического материала менее драматичен по своим последствиям, чем его недостаток. Вместе с тем сходство клинических проявлений для многих хромосомных болезней свидетельствует как о большом числе генов, контролирующих развитие каждого органа и ткани, разбросанных по геному, так и о сложной системе связей между развивающимися зачатками разных систем организма.
Далее мы кратко опишем только наиболее частые хромосомные болезни человека, отсылая интересующихся этой проблемой читателей к руководствам по клинической генетике.
Трисомии. Самой частой из трисомий и вообще одной из самых частых наследственных болезней является трисомия 21, или синдром Дауна. Цитогенетическая природа синдрома Дауна была установлена Ж.Леженом в 1959 г. Синдром встречается в среднем с частотой 1 на 700 живорожденных, но частота синдрома зависит от возраста матерей и повышается с его увеличением. У женщин старше 45 лет частота рождения больных с синдромом Дауна достигает 4 %. Основные клинические проявления синдрома Дауна представлены в табл. 8.3.
Таблица 8.3. Основные клинические проявления синдрома Дауна
Симптомы |
Частота встречаемости, % |
Умственная отсталость |
99 |
Плоское лицо |
90 |
Монголоидный разрез глаз |
80 |
Эпикант |
40 |
Пятна Брушфильда на радужке |
50 |
Косоглазие |
60 |
Аномалии ушных раковин |
50 |
Высокое или готическое небо |
70 |
Брахицефалия |
75 |
Плоский затылок |
78 |
Мелкие зубы |
65 |
Короткая широкая шея |
45 |
Большой язык |
50 |
Врожденный порок сердца |
8 |
Дуоденальная обструкция |
70 |
Короткие конечности |
70 |
Широкие короткие кисти, короткие пальцы |
70 |
Единственная ладонная складка |
20 |
Сандалевидная щель |
45 |
Гипотония |
60 |
Низкий рост |
80 |
Цитогенетическими причинами синдрома Дауна являются регулярная трисомия — 95 %, транслокации хромосомы 21 на другие хромосомы — 3 % и мозаицизм — 2 %. Молекулярногенетические исследования позволили выявить критический район хромосомы 21, ответственный за основные клинические проявления синдрома Дауна, — 21q22.
Повторный риск при регулярной трисомии 21 составляет примерно 1:100 и зависит от возраста матери. При семейной транслокации показатели риска варьируют от 1 до 3 %, если носителем транслокации является отец, и от 10 до 15 %, если носителем транслокации является мать. Как уже отмечалось, при редких случаях транслокации 21q21q повторный риск составляет 100 %.
Трисомия 18, или синдром Эдвардса, встречается значительно реже, чем трисомия 21. Частота синдрома составляет примерно 1 на 5000 живорожденных, у девочек он наблюдается примерно в 3 раза чаще, чем у мальчиков. Клинические проявления синдрома Эдвардса значительно более тяжелые, чем синдрома Дауна, обычно больные погибают на первых неделях жизни. Фенотипические признаки трисомии 18 приведены в табл. 8.4.
Таблица 8.4. Основные клинические проявления синдрома Эдвардса
Симптомы |
Частота встречаемости, % |
Тяжелая задержка психомоторного и физического развития |
100 |
Затруднения при глотании, проблемы с кормлением |
100 |
Низкая масса тела при рождении |
100 |
Гипертонус |
65 |
Пороки развития головного и спинного мозга |
30 |
Менингомиелоцеле |
15 |
Выступающий затылок |
90 |
Низко посаженные, уродливые уши |
90 |
Птоз, эпикант, микрофтальмия |
30 |
Расщелина губы и неба |
15 |
Микрогнатия |
90 |
Короткая шея с избыточностью кожи |
60 |
Короткая грудина |
90 |
Врожденный порок сердца (обычно дефект межжелудочковой перегородки) |
95 |
Эвентерация диафрагмы |
30 |
Паховая и пупочная грыжи |
60 |
Пилоростеноз |
30 |
Симптомы |
Частота встречаемости, % |
Пороки развития почек |
60 |
Крипторхизм |
100 |
Косолапость |
50 |
Сгибательная деформация пальцев, перекрест пальцев рук |
90 |
Частичная синдактилия |
30 |
Гипоплазия ногтей на руках и ногах |
30 |
Короткие изогнутые большие пальцы ног |
60 |
При цитогенетическом исследовании обычно обнаруживают регулярную трисомию 18. Как и при синдроме Дауна, выявляется связь между частотой трисомии 18 и возрастом матери. В большинстве случаев дополнительная хромосома имеет материнское происхождение. Около 10 % трисомии 18 обусловлены мозаицизмом или несбалансированными перестройками, чаще робертсоновскими транслокациями.
Трисомия 13, или синдром Патау, встречается с частотой 1 на 10 ООО новорожденных. Клинические проявления синдрома Патау, так же как и синдрома Эдвардса, как правило, очень тяжелые и включают множественные врожденные пороки развития (табл. 8.5). Смертность среди новорожденных с синдромом трисомии 13 в первые недели жизни очень высока.
Таблица 8.5. Основные клинические проявления синдрома Патау
Симптомы |
Частота встречаемости, % |
Глубокая задержка умственного и физического развития |
100 |
Микроцефалия |
70 |
Предположительно глухота |
70 |
Гипотония |
45 |
Судороги |
45 |
Дефекты скальпа |
30 |
Гипертелоризм |
90 |
М икрофтал ьмия |
65 |
Эпикант |
65 |
Отсутствие бровей |
30 |
Колобома радужки |
30 |
Низко посаженные, уродливые уши |
90 |
Расщелина губы и/или неба |
65 |
Короткая шея |
65 |
Врожденный порок сердца (ДМЖП, ДМПП, коарктация аорты) |
65 |
Симптомы |
Частота встречаемости, % |
Единственная пупочная артерия |
30 |
Паховые и пупочная грыжи |
30 |
Омфалоцеле |
15 |
Капиллярная гемангиома |
65 |
Полидактилия |
65 |
Расщелина кистей |
20 |
Косолапость |
20 |
Аномалии почек |
90 |
Цитогенетически обычно выявляется регулярная трисомия 18, повторный риск для которой низкий. Изредка встречаются случаи 13-D робертсоновских транслокаций. Риск для носителей таких транслокаций повторных случаев трисомии 18 у потомства существенно ниже, чем при синдроме Дауна, и составляет менее 1 %.
Еще реже, чем трисомии 13 и 18, встречаются полные или частичные трисомии по другим аутосомам, в частности по хромосомам 8, 9, lq, 2р, 2q, 3q, 5р и т.д. Практически все они проявляются множественными врожденными пороками развития.
Трисомии или в более общем виде полисомии по половым хромосомам встречаются почти так же часто, как и трисомия по хромосоме 21. Кариотипы с дополнительными хромосомами X и Yпредставлены в табл. 8.6.
Таблица 8.6. Полисомии по половым хромосомам
Тип хромосомных нарушений |
Частота, % |
Частота в популяции |
Синдром Клайнфе |
л т е р а |
1 на 1000 мужчин |
47,ХХУ |
82 |
|
48,XXXY |
3 |
|
49XXXXY |
<1 |
|
Мозаики |
8 |
|
Прочее |
6 |
|
Полисомия Jf |
|
1 на 1000 женщин |
47,ХХХ |
>98 |
|
48,ХХХХ |
Редко |
|
49,ХХХХХ |
Редко |
|
Мозаики |
Редко |
|
Полисомия Y |
|
1 на 1000 женщин |
47,XYY |
>98 |
|
Прочее |
Редко |
|
Как видно из табл. 8.6, наличие хромосомы Yпри синдроме Клайнфелтера независимо от числа дополнительных хромосом X определяет развитие мужского пола. Самым частым является кариотип 47,XXY(табл. 8.7).
Таблица 8.7. Основные клинические проявления синдрома Клайнфелтера при кариотипе 47,XXY
Симптомы |
Частота встречаемости, % |
Высокий рост, астеничное телосложение |
80 |
Умственная отсталость |
5 |
Маленькие наружные половые органы |
50 |
Гистологические доказательства нарушений сперматогенеза |
100 |
Гинекомастия |
55 |
Сниженный уровень тестостерона |
80 |
Повышенный уровень гонадотропина |
75 |
Плохой рост волос на лице |
80 |
Клинические проявления синдрома Клайнфелтера усиливаются с увеличением числа хромосом X в кариотипе. Из мозаичных кариотипов самым частым является 46,XY/47,XXY. Встречаются также мозаики 46,XX/47,XXY; 46,XY/47,XXY/
XXYY и 47,XXY/48,XXYY.
У женщин с дополнительными хромосомами X, число которых может доходить до 4, клинические проявления синдрома полисомии по хромосоме X могут либо вовсе отсутствовать, либо проявляться небольшой умственной отсталостью. Такие женщины, как правило, фертильны, а кариотип их потомства обычно нормальный.
Мужчины с кариотипом XYY встречаются относительно часто. Клинических проявлений этот кариотип не имеет, но считают, что мужчины XYY более высокого роста, чем в среднем в популяции, и более агрессивны.
Моносомии. Моносомия у человека известна только в отношении хромосомы X. Общее название для разных типов моносомий по хромосоме X — синдром Шерешевского—Тернера (частота в популяции 1 на 1000 женщин). Цитогенетическое разнообразие, объединяемое под этим эпонимом, представлено в табл. 8.8.
Женщины с синдромом Шерешевского—Тернера имеют достаточно характерный фенотип, который включает симптомы, указанные в табл. 8.9.
Синдром Шерешевского—Тернера возникает не только при полной, но и частичной моносомии по хромосоме X,
Таблица 8.8. Цитогенетические нарушения при синдроме Шере- шевского—Тернера
Тип хромосомных нарушений |
Частота, % |
45,X |
57 |
46,X,i(Xq) и мозаики с линиями клеток i(Xq) |
17 |
46,X,del(Xq) и мозаики с линиями клеток del(Xq) |
1 |
Мозаики 45,Х/46,XX; 45,X/47,XXX |
12 |
Мозаики 45,Х/46,XY |
4 |
Прочее [del(Xp), г(Х), мозаики] |
9 |
Таблица 8.9. Основные клинические проявления синдрома Ше- решевского—Тернера
Симптомы |
Частота встречаемости, % |
Низкий рост |
97 |
Первичная аменорея |
96 |
Стерильность |
70 |
Лимфатический отек кистей и стоп при рождении |
40 |
Крыловидные складки на шее |
53 |
Пороки сердца |
20 |
Пороки развития почек |
40 |
Умственная отсталость |
18 |
Высокое небо |
45 |
Широкая грудная клетка, часто с деформацией |
40 |
Cubitus valgus |
48 |
Снижение слуха |
53 |
когда отсутствует длинное или короткое плечо этой хромосомы. Значительный процент случаев синдрома обусловлен мо- заицизмом. Основные типы мозаичных кариотипов при синдроме Шерешевского—Тернера представлены в табл. 8.8.
Моносомики по аутосомам нежизнеспособны. Однако описаны, а в некоторых случаях достаточно хорошо изучены частичные моносомии или делеции и обусловленные ими хромосомные болезни.
Делеция короткого плеча хромосомы 4 (синдром 4р, или синдром Вольфа—Хиршхорна). Впервые эта делеция была описана в 1965 г. Ее клинические проявления приведены в табл. 8.10.
Симптомы |
Частота встречаемости, % |
Глубокая умственная отсталость |
100 |
Микроцефалия |
91 |
Затруднения при глотании |
76 |
Дефект скальпа по средней линии |
14 |
Низкая масса тела при рождении |
89 |
Судороги |
47 |
Гипертелоризм |
74 |
Колобома радужки |
31 |
Антимонголоидный разрез глаз |
74 |
Преаурикулярные синусы, или выросты |
33 |
Расщелина губы и/или неба |
57 |
Гипоспадия или крипторхизм у мальчиков, гипоплазия матки у девочек |
64 |
Крючковатый нос |
64 |
Низко посаженные, уродливые уши |
69 |
Пороки сердца |
55 |
Косолапость |
70 |
Микрогнатия |
55 |
«Обезьянья» складка |
70 |
Делеция короткого плеча хромосомы 5 (синдром 5р-, или синдром «кошачьего крика»). Хромосомная природа синдрома 5р- была установлена Ж.Леженом с сотрудниками в 1963 г. у новорожденного с задержкой развития, микроцефалией и своеобразным плачем, похожим на крик кошки. Клинические проявления синдрома 5р- имеют много общего с синдромом 4р- (табл. 8.11).
С возрастом фенотип больных существенно меняется. Больные обычно низкого роста, у них удлиненное лицо, нередко асимметричное, плохое развитие мышечной системы, сколиоз, преждевременное поседение и неправильный прикус.
Приблизительно 85 % всех случаев синдрома являются спорадическими, 15 % наследуется от фенотипически нормальных родителей, носителей сбалансированной хромосомной перестройки (транслокации или инверсии).
Делеции короткого плеча всех акроцентрических хромосом практически не имеют каких-либо серьезных клинических проявлений.
Как отдельные нозологические формы (синдромы) описаны делеции 18р, 18q, 21q и 22q.
Симптомы « |
Частота встречаемости, % |
Низкая масса тела при рождении |
100 |
Умственная отсталость |
100 |
Затруднения при глотании |
30 |
Плач, похожий на крик кошки |
100 |
Дыхательный стридор |
60 |
Ларингомаляция |
20 |
Микроцефалия |
90 |
Гипертелоризм |
70 |
Косоглазие |
50 |
Антимонголоидный разрез глаз |
50 |
Низко посаженные, уродливые уши |
60 |
Микрогнатия |
60 |
«Обезьянья» складка |
70 |
В ядре каждой соматической клетки организма человека в норме содержится 46 хромосом. Набор хромосом каждого индивидуума, как нормальный, так и патологический, называют кариотипом. Из 46 хромосом, составляющих хромосомный набор человека, 44 или 22 пары представляют аутосомные хромосомы, последняя пара — половые хромосомы. У женщин конституция половых хромосом в норме представлена двумя хромосомами X, у мужчин — X и Y. Хромосомы одной пары называют гомологами, или гомологичными хромосомами. В половых клетках (сперматозоидах и яйцеклетках) содержится гаплоидный набор хромосом, т.е. 23 хромосомы.
В каждой хромосоме выявляется перетяжка, которую называют центромерой. По положению центромеры хромосомы классифицируют на метацентрические, акроцент- рические и субметацентрические.
Материал, из которого построены хромосомы, называют хроматином. Он состоит из ДНК и окружающих ее ги- стонов и других белков. Ту часть хроматина, которая слабо окрашивается специальными красителями для хромосом, называют эухроматином, а ту, которая окрашивается интенсивно, — гетерохроматином. Считают, что эухрома- тиновые районы хромосом содержат активно экспрессирующиеся гены, гетерохроматиновые районы, напротив, содержат неактивные гены и неэкспрессирующиеся повторяющиеся последовательности ДНК.
Соматическая клетка может находиться в двух состояниях — интерфазе и делении. Смену этих состояний друг другом Называют клеточным циклом. Во время интерфазы клетка удваивает свое содержимое, включая хромосомы. Интерфазу принято делить на три стадии: Gl, S и G2. На стадии G1 в клетке происходит синтез РНК, белков и осуществляется весь сложный комплекс метаболизма. На стадии S происходит репликация ДНК. На стадии G2 исправляются ошибки, допущенные при репликации ДНК, и происходят другие процессы, подготавливающие клетку к митозу.
Процесс деления соматических клеток, во время которого также происходит деление ядра, называют митозом. До вхождения клетки в митоз каждая хромосома представлена двумя идентичными нитями, которые являются результатом репликации ДНК во время фазы синтеза клеточного цикла. Эти нити называют хроматидами. Во время деления ядра клетки хроматиды каждой хромосомы расходятся в две вновь возникшие клетки. Таким образом, в соматических клетках на протяжении всей жизни человека сохраняется одно и то же число хромосом и, следовательно, все соматические клетки генетически идентичны друг другу.
Митоз принято делить на отдельные стадии (или фазы): профазу, прометафазу, метафазу, анафазу и телофазу.
Мейозом называют процесс деления ядер зародышевых клеток при их превращении в гаметы. Мейоз включает два деления клеток, которые называют соответственно мейозом I и мейозом И. Каждое из этих делений формально состоит из тех же стадий, что и митоз: профазы, метафазы, анафазы и телофазы. Мейоз I называют также редукционным делением, так как в результате этого деления число хромосом во вновь образующихся клетках уменьшается в 2 раза. Мейоз II по механизму сходен с обычным митозом, но митотически делится удвоенный гаплоидный набор хромосом. В результате второго мейотического деления образуются в мужском гаметогенезе две спермати- ды, а в женском гаметогенезе — яйцеклетка, так как из второй дочерней клетки образуется так называемое направительное тельце. Мейоз объясняет многие генетические феномены, в том числе менделевские правила наследования.
Различают два основных типа хромосомных мутаций — численные и структурные. Численные мутации делятся на анэуплоидии, когда мутации выражаются в утрате или появлении дополнительной одной либо нескольких хромосом, и полиплоидии, когда увеличивается число гаплоидных наборов хромосом. Потерю одной из хромосом называют моносомией, а возникновение дополнительного гомолога у пары хромосом — трисомией. Обычно трисомии возникают в результате нарушения расхождения гомологичных хромосом в анафазе мейоза I. В результате в одну дочернюю клетку попадают обе гомологичные хромосомы, а во вторую дочернюю клетку не попадает ни одна из хромосом бивалента. Иногда, однако, трисомия может быть результатом нарушения расхождения сестринских хрома- тид в мейозе II. В этом случае в одну гамету попадают две совершенно одинаковые хромосомы, что при оплодотворении нормальным спермием даст трисомную зиготу. Данный тип хромосомных мутаций, ведущих к трисомии, называют нерасхождением хромосом.
Структурные хромосомные мутации представлены транслокациями (реципрокными и робертсоновскими), делециями, инсерциями, инверсиями (парацентрическими и перицентрическими), кольцами и изохромосомами. Структурные мутации хромосом могут возникать только в результате разрыва хромосом с последующим воссоединением, сопровождающимся нарушением исходной конфигурации хромосом. Структурные хромосомные мутации могут быть сбалансированными или несбалансированными. При сбалансированных мутациях нет утраты или избытка генетического материала, поэтому они не имеют фенотипических проявлений, кроме тех случаев, когда в результате разрыва хромосомы в месте разрыва оказывается функционально важный ген. У носителей сбалансированных хромосомных мутаций могут образовываться несбалансированные по хромосомному набору гаметы в результате нарушений в распределении хромосомного материала в мейозе, и как следствие этого у плода, возникшего от оплодотворения такой гаметой, хромосомный набор окажется также несбалансированным. При несбалансированном хромосомном наборе у плода развиваются тяжелые клинические проявления патологии, как правило, в виде комплекса врожденных пороков развития.
При численных аномалиях как половых хромосом, так и аутосом нередко обнаруживают мозаицизм, проявляющийся одновременным существованием в организме как эуплоидных, так и анэуплоидных клеток в разных пропорциях. Мозаицизм может возникнуть вследствие митотического нерасхождения или из-за потери одной из хромосом в результате анафазного отставания.
Недавно установлено, что причиной ряда синдромов, тип наследования которых оставался неизвестным, так как они встречались почти всегда в виде изолированных
случаев, являются микроделеции. Самыми известными и частыми наследственными синдромами, обусловленными микроделециями района 15qll—ql3, являются синдром Прадера—Вилли и синдром Энжельмена.
После того как в 1956 г. были разработаны методы анализа хромосом человека, за короткое время была установлена хромосомная природа многих заболеваний, в том числе синдрома Дауна (47,XX/XY, +21), синдрома Клайнфелтера (47,XXТ), синдрома Шерешевского—Тернера (45,X) и некоторых других синдромов аутосомных трисо- мий. В настоящее время различают почти 1000 хромосомных синдромов, их вклад в спонтанные аборты, неонатальную смертность и заболеваемость весьма значительный. Большинство хромосомных болезней характеризуется наличием множественных врожденных пороков развития.