

Наследование, сцепленное
С ХРОМОСОМОЙ Y
Хромосома Y содержит относительно небольшое число генов. К началу 2002 г. в ней картировано немногим более 35 генов, из них только 7 вызывают наследственные болезни, в том числе пигментный ретинит, несколько форм азооспермии, дисхондростеоз и гонадобластому, нарушение диффе- ренцировки пола и др. & хромосоме Y находится несколько генов «домашнего хозяйства», гомологичные им гены расположены в хромосоме X. Последние гены не инактивируются при инактивации хромосомы X.
Как следует из табл. 5.5, хромосома У передается только от отца к сыну, и такое наследование называют голандриче- ским. Убедительно доказать, что признак наследуется сцеп- ленно с хромосомой Y по родословным крайне трудно, так его надо дифференцировать от аутосомно-доминантного на* следования. Только большие семьи, в которых среди сибсов встречаются и девочки, и мальчики, но признак такой же, как у отца, есть только у мальчиков, позволяют заподозрить, что речь идет об У-сцепленном наследовании.
При У-сцепленном заболевании больны будут также только мужчины, но в отличие от А-сцепленной патологии пораженный отец будет передавать заболевание только своим сыновьям, его дочери и их дети всегда здоровы.
Генетические механизмы определения пола
У человека важнейшим фактором определения пола в эмбриональном развитии является хромосома Y. Далее, в главе, посвященной хромосомам человека, этот вопрос будет рассмотрен более подробно. Сейчас же мы упомянем, что независимо от числа хромосом X отсутствие хромосомы Y в ка- риотипе развивающегося организма не позволяет дифференцироваться мужскому полу.
Относительно недавно было показано, что в дифференциации мужского пола особая роль принадлежит гену SRY (сокращение расшифровывается как «пол-определяющая область хромосомы Y). Мутации в гене SRY приводят к тому, что у лиц с кариотипом XY развивается женский фенотип. При введении гена SRY в клетки эмбриона мыши женского пола происходит трансформация пола и у новорожденного мышонка пол оказывается мужским. Ген SRY расположен в области YplL3, т.е. в коротком плече хромосомы У, на границе с псевдоаутосомной областью этой хромосомы.
Ранние эмбриональные гонады могут дифференцироваться как в яички, так и в яичники. Предшественником гонад у млекопитающих является половой гребешок. Он возникает из мезонефроса и целомического эпителия. Половые клетки возникают из примитивной полоски и затем мигрируют в гонады. Эти клетки бипотенциальны. Если половые клетки попадают в тестикулярную среду и контактируют с клетками Сертоли (сустентоцитами), то дифференцируются в сперма- тогонии. Если же контакта с сустентоцитами не происходит, то половые клетки дифференцируются в оогонии. Другими полоспецифическими органами являются мезонефральные и парамезонефральные протоки1. Оба типа протоков имеются у эмбрионов независимо от их хромосомной конституции. Рост и дифференцировка протоков зависят от гормонов, секрети- руемых эмбриональными яичками. Тестостерон, который декретируется клетками Лейдига (гландулоцитами) яичек, вызывает дифференциацию мезонефральных протоков в предстательную железу, семенные пузырьки и семявынося-
Jno старой классификации соответственно вольфов и мюллеров протоки.
Регрессия
парамезо-
нефральных
протоков
Рост
семенных полового члена и влагалища
пузырьков
и предстательной
семявыносящих железы
протоков
Рис.
5.7. Дифференцировка
пола.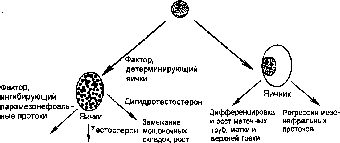
Эмбриональные гонады могут дифференцироваться в яички и яичники. Если эмбриональные гонады находятся у плода мужского пола, то ген SRY, локализованный в Y хромосоме, продуцирует фактор, детерминирующий дифференцировку эмбриональных яичек. Эмбриональные яички секретиру- ют гормоны, определяющие развитие мужских половых органов. В отсутствие гена SRY эмбриональные гонады дифференцируются в яичник (из: Osier Н. Sex determination: lessons from families and embryos/Clin.Genet. — 2001. — Vol. 59. - P. 207-215).
щие протоки. При отсутствии сигнала тестостерона эти протоки регрессируют, а парамезонефральные протоки дифференцируются в матку, маточные трубы и верхнюю треть вагины. У эмбрионов мужского пола сустентоциты яичек секретируют гормон — фактор ингибиции парамезонефраль- ных протоков, который вызывает регресс этих протоков (рис. 5.7).
Ген SRY экспрессируется в половом гребешке в момент индукции дифференциации яичек. В случае делеций или иных мутаций в гене SRYяички не дифференцируются. Продукт гена SRY является, вероятно, ядерным фактором транскрипции, связывая белки, модулирующие транскрипцию. Белок SRY содержит HMG-бокс, который связывается с консенсусной последовательностью ААТААЦ ДНК. Примерно у 20 % 46,АХ-мужчин ген SRY не обнаруживают, что позволяет предположить его неабсолютную обязательность для мужской дифференциации пола.
Установлено, что, кроме гена SRY\ несколько аутосомных генов играют определенную роль в мужской дифференциации пола. Один из них — ген супрессор опухоли Вильмса, кодирует фактор транскрипции, связывающийся со специфи-
оо
os
Эмбриональный период Плодный период
44-й ДПО С18С Яички
SF1
DAXI
Начало
образования
половой хорды /
WT1 SF1
DAX1
32-й ДПО С14С
SRY
SOX9
WT1
Сустентоциты
WT1
Сустентоциты
SF1 DAX1
I
WT1
SF1
DAX1
WT1
SF1
WT1
Гландулоциты
I
i SF1 I I
полового гребешка гонады
WT1
i WT1
| SF1 Отсутствие J DAX1 начало
; ► очевидной ! фолликулогенеза
дифференцировки дифференцировки i i
i i
DAX1
Отсутствие
очевидной
DAX1
Яичник
Мезонефроз
Пролиферирующий целомический — эпителий
SF1
Надпочечникогонадный
зачаток
Недифференцированные
состояния
42-й ДПО С17С
SRY
SOX9
Недиф.
Образование
i Гландулоциты
52-й ДПО С21С
SRY
SOX9
ческими ДНК-последовательностями. Точковые мутации в этом гене иногда приводят к формированию гонадно противоположного пола. Второй ген такого же типа — SOX9. Белок, кодируемый этим геном, также содержит HMG-бокс. Мутации в гене SOX9 вызывают кампомелическую дисплазию, при которой у индивидуумов 46,XY фенотипически развивается женский пол. Стероидогенный фактор 1 (SF1), кодируемый соответствующим геном, является фактором транскрипции, регулирующим экспрессию ряда генов, которые участвуют в продукции стероидных гормонов и мужской половой дифференциации. Показано, что гемизиготная мутация в этом гене ассоциирует с надпочечниковой недостаточностью и гонадным дисгенезом у лиц с кариотипом 46,XY. Ген MIS, картированный в области 19р13, отвечает за синтез фактора, ингибирующего парамезонефральные протоки. Мутации в этом гене ведут к проявлениям синдрома персистен- ции указанных протоков. Экспрессия MIS регулируется продуктами генов S0X9 и SF1.
Для нормального развития гонад необходимы также Х-хро- мосомные локусы. У некоторых лиц с гонадным дисгенезом 46,АТ наблюдалась дупликация области Хр21. В этой области находится ген DAX1, белок которого функционирует как транскрипционный фактор. Еще один ген в хромосоме X — ХН2, играет не вполне выясненную роль в дифференцировке яичек. Он кодирует фермент геликазу, которая расплетает ДНК, делая ее доступной для транскрипционных факторов.
<
Рис. 5.8. Экспрессия генов, определяющих пол во время развития плода у человека.
Вверху показаны стадии дифференциации гонад. Ген хромосомы У, определяющий пол, названный 57?У, экспрессируется в половом гребешке в момент индукции дифференциации яичек. Продукт гена SRY (фактор, детерминирующий яички), вероятно, является ядерным фактором транскрипции, связывая белки, модулирующие транскрипцию. Еще несколько аутосомных генов играют определенную роль в мужской дифференциации пола. Один из них — ген-супрессор опухоли Вильмса, который кодирует фактор транскрипции, связывающийся со специфическими ДНК-последовательностями (WTJ). Второй ген такого же типа — SOX9. Он контролирует синтез белка, который называется SRУ-подобным белком 9, содержащим HMG-бокс. Стероидогенный фактор ](SF1) является фактором транскрипции, регулирующим экспрессию ряда генов, которые участвуют в продукции стероидных гормонов и мужской половой дифференциации. Для нормального развития необходимы также Л'-хромосомные локусы. К ним относятся ген DAX1 (дозозависимая инверсия пола, врожденная гипоплазия надпочечников и хромосома X и ген ХН2). Ген ХН2 кодирует геликазу, которая раскручивает ДНК, делая ее доступной для транскрипционных факторов. Несколько детерминирующих генов взаимодействуют, регулируя экспрессию гена антимюллерова гормона (MIS или MIF). К ним относятся SOX9 и SF1 (из: Oster Н. Sex determination: lessons from families and embryos/Clin. Genet. — 2001. — Vol. 59. — P. 207—215). ДПО — дни после оплодотворения; CXXC — стадии такого-то числа сомитов.
Нет сомнений в том, что современные представления о генетическом контроле становления пола далеко не полные. Это относится не только к тому, какие гены вовлечены в данный процесс, но и к тому, на каких стадиях развития и в каких тканях эти гены проявляются. На примере генетического контроля определения пола мы сталкиваемся с некоторыми кардинальными проблемами генетики развития — ткане- и времяспецифичности действия генов. При образовании полового гребешка из мезонефроса и целомического эпителия в соответствующих тканях обнаружена активность гена WT1, а в зачатке надпочечника, который также имеет отношение к образованию полового гребешка, — генов SF1 и DAX1. Половой гребешок формирует недифференцированные гонады при участии тех же генов. В предшественниках половых клеток эмбриона мужского пола резко возрастает активность генов SRYи S0X9. Практически все перечисленные гены и еще ген MIS активны в сустентоцитах, а гены SF1 и DAX1 — в гландулоцитах. В то же время у женского эмбриона вплоть до плодного периода гонады остаются недифференцированными, хотя в них обнаруживают активность генов WT1, SF1 и DAX1 (рис. 5.8).
ПРИМЕРЫ РАЗЛИЧНЫХ КЛАССОВ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
В этом разделе мы приведем в табличной форме примеры для некоторых групп наследственных менделирующих заболеваний, успешного изучения их молекулярно-генетической природы. В таблицы включены преимущественно заболевания, гены которых клонированы и известны белок или поли- пептидная цепь, структуры которых определяются соответствующим геном. Это далеко не все менделирующие наследственные заболевания, гены которых клонированы. Таких заболеваний уже насчитывается больше тысячи, их число увеличивается очень быстрыми темпами. Однако даже те данные, которые будут приведены в этой главе, показывают, насколько глубоко современный врач должен знать биологическую основу жизнедеятельности человека, чтобы он мог представить себе, как формируется патогенез заболеваний на молекулярном уровне.
Наследственные болезни обмена веществ
Наследственные болезни обмена веществ являются предметом одного из разделов биохимической генетики человека, которую можно рассматривать как генетическую основу всех метаболических превращений, осуществляемых в организме человека. Поскольку, однако, этот учебник посвящен проблемам медицинской генетики, то мы сочли целесообразным ограничиться рассмотрением только наследственных болезней обмена веществ.
В главе, посвященной истории медицинской генетики, указывалось, что одним из выдающихся моментов этой истории было открытие А.Гэрродом в начале XX в. биохимической природы наследственных болезней обмена веществ, которые сам Гэррод назвал врожденными ошибками метаболизма.
Долгое время к указанным болезням относили только такие нарушения метаболизма, которые были связаны с недостаточностью ферментов, т.е. в полном согласии с гипотезой Бидла и Тэтума «один ген — один фермент». Однако позднее, когда эта гипотеза претерпела определенную эволюцию, связанную с более точным определением функции генов, в число наследственных болезней обмена стали включать также и другие наследственные болезни, обусловленные недостаточностью структурных, транспортных белков, белков каналов и рецепторов, белков иммунной защиты и т.д. При таком подходе большая часть моногенных, или менделирующих, наследственных болезней оказывается включенной в наследственные болезни обмена веществ. Однако это делается не совсем последовательно. В результате одни авторы считают, что известно 200—300 наследственных болезней обмена веществ, а другие полагают, что к настоящему времени уже описано более 600 таких болезней.
Изучение наследственных болезней обмена веществ оказалось полезным не только с чисто медицинской позиции, поскольку были расшифрованы биохимические и молекулярные основы патогенеза соответствующих заболеваний, но и с общебиологических позиций, так как при этом выявлялись и уточнялись особенности метаболических путей и их отдельных звеньев у человека. Представление о разнообразии наследственных болезней обмена веществ дает табл. 5.7.