

Механизмы аутосомной доминантности
В предыдущих главах мы рассмотрели, что «делает» ген и как нарушается его функция в случае возникновения мутаций. Однако молекулярные механизмы действия гена или нарушения его функции, которые могут быть описаны как утрата функции либо усиление функции, не могут объяснить явление доминантности. Это связано с тем, что мы рассматривали функцию гена как такового, а доминантность и рецессивность являются результатом взаимодействия как минимум разных аллелей одного гена.
Уже долгое время существует представление о том, что аутосомно-рецессивные заболевания обусловлены недостаточностью ферментов, а аутосомно-доминантные — недостаточностью структурных белков.
Действительно, многие рецессивные заболевания обусловлены недостаточностью ферментов. Из этого следует, что нормальные аллели генов многих ферментов являются доминантными и их продукты, обеспечивающие 50 % нормальной ферментативной активности, достаточны для обеспечения нормального функционирования клеток организма (такие аллели называют гаплодостаточными). Рецессивные аллели таких генов можно характеризовать как аллели с потерей функции. Это становится очевидным для гомозигот по рецессивным аллелям генов многих ферментов человека: ферментативная активность при многих наследственных болезнях обмена веществ не превышает 10 %.
Теперь, однако, известно, что не только недостаточность ферментов, обусловливающая наследственные болезни обмена веществ, наследуется аутосомно-рецессивно. При таком частом аутосомно-рецессивном наследственном заболевании, как муковисцидоз (мы его уже рассматривали в связи с молекулярными механизмами мутаций), нарушается функция белка хлорного канала, при спинальных амиотрофиях, которые также наследуются аутосомно-рецессивно, меняется белок гемин, блокирующий апоптоз, при (3-талассемии, частом аутосомно-рецессивном заболевании во многих регионах мира, нарушаются синтез (3-цепей глобина и последующая сборка гемоглобина взрослых, представляющего собой мультимер, состоящий из двух а- и двух (3-цепей глобина. Можно привести еще много примеров, когда рецессивно наследуются наследственные заболевания, при которых мутации происходят в генах, контролирующих синтез неферментных белков. Однако известны мутации в генах, контролирующих синтез ферментов, что вызывает аутосомно-доминантные заболевания. Так происходит, в частности, при 5 формах наследственной доминантной порфирии, обусловленных дефектами 5 разных ферментов. Для одной из форм порфирии — острой перемежающейся порфирии — показано, что доминантный эффект является результатом недостаточного ингибирования синтетазы 5-аминолевуленовой кислоты гемом, образование которого нарушено из-за мутационного дефекта порфобилиногендезаминазы (собственно мутантного белка). Таким образом, доминантный эффект как бы вторичен по отношению к мутационно измененному гену порфобилиногендезаминазы.
Доминантность нередко возникает, когда мутантная поли- пептидная цепь должна вступить во взаимодействие с нормальной полипептидной цепью, синтез которой контролируется нормальным аллелем того же гена, или полипептидными цепями, синтез которых контролируется другими генами при образовании молекулы зрелого мультимерного белка. В результате все или какая-то часть молекул оказываются дефектными, что и создает эффект доминантности. Ярким примером доминантных заболеваний такого рода являются различные формы несовершенного остеогенеза. Большинство случаев несовершенного остеогенеза обусловлены дефектом коллагена I типа, молекула которого состоит их трех субъединиц. Две из них, обозначаемые как npoal(I), кодируются геном, расположенным на хромосоме 17. Третья цепь — проа2(1) — кодируется геном, расположенным на хромосоме 7. Три цепи проколлагена I типа ассоциируют друг с другом, образуя сложную трехспиральную молекулу белка. Мутации в генах любой из цепей коллагена I типа будут приводить к нарушению образования зрелой молекулы коллагена и фибрилл, построенных из этого белка. Подобного рода мутации называют доминантно негативными.
Следует заметить, что при доминантных заболеваниях, которые обычно бывают проявлением гетерозиготного носите- льства мутантного гена, нормальный аллель продолжает обеспечивать синтез 50 % нормальных полипептидных цепей, но этого недостаточно для обеспечения нормальной функции зрелой белковой молекулы. Поэтому такие нормальные аллели называют гаплонедостатонными.
НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С ХРОМОСОМОЙ X
Число известных Лг-сцепленных заболеваний, конечно, меньше, чем доминантных или рецессивных, гены которых «разбросаны» по 22 аутосомам. Тем не менее известно около 300 генов, локализованных в хромосоме X, вызывающих наследственные болезни (гены, локализованные в хромосоме X, называют Л'-сцепленными). К ним относятся гены гемофилии А, миопатии Дюшенна, ЛГ-сцепленного ихтиоза, пигментной дистрофии сетчатки, ангидротической формы эктодермальной дисплазии, синдрома ломкой хромосомы X с умственной отсталостью, гидроцефалии, синдромов Коффина — Лоури, Пейна, Опитца, одной из форм мукополисахаридоза, Х-сцеп- ленной невральной амиотрофии, недостаточности глюко- зо-6-фосфатдегидрогеназы и многих других заболеваний.
Прежде чем мы перейдем к изложению материала о наследовании, сцепленного с хромосомами X и У, и особенностях родословных при этих типах наследования, напомним, что у человека пол гетерогаметен. Женщины имеют во всех клетках две хромосомы X, а мужчины — одну X и одну Y хромосомы. Хромосома Y не гомологична хромосоме X, в ней содержится небольшое число генов. Мужчины являются гемизиготами по хромосоме X и всем содержащимся в ней генам. Наследование половых хромосом происходит как наследование простых менделевских признаков (табл. 5.5; это уже привычная решетка Пеннета).
|
|
Гаметы |
матери |
|
|
*1 |
*2 |
Гаметы отца |
X |
ХхХ |
х2х |
|
Y |
Х\ у |
Х2 Y |
Из табл. 5.5 видно, что при случайном объединении гамет должно образовываться равное количество зигот женского и мужского пола.
Поведение генов, которые расположены в половых хромосомах, строго соответствует поведению самих хромосом. Если мутантный ген находится в одной из хромосом X матери (например, в хромосоме Х\)> то эту хромосому X получат 50 % сыновей и 50 % дочерей матери-носительницы. Если ген «отвечает» за рецессивное заболевание, то сама мать должна быть здорова, так как у нее есть вторая нормальная хромосома Х9 но оно разовьется у той половины сыновей, которые получили хромосому Х\ с мутантным геном, поскольку хромосома Y не гомологична хромосоме 1,ив ней просто не содержится пар абсолютному большинству генов хромосомы X. У дочерей, получивших от матери измененную хромосому, заболевание также не разовьется, так как они получат нормальную вторую хромосому X от отца. Таким образом, если мать является носительницей ^-сцепленного рецессивного гена, риск быть больным у ее сыновей составит или 50 %, а у ее дочерей — 0 %. В то же время риск быть гетерозиготными носительницами у дочерей равен 50 %.
Для рецессивных X-сцепленных заболеваний характерно, как это следует из изложенного, что поражаться ими будут мужчины, родственники друг другу по материнской линии. Это могут быть двоюродные братья, матери которых родные сестры, или дяди и племянники больного по материнской линии и т.д. (рис. 5.5).
В том случае, если отец все-таки болен, все его сыновья будут здоровы, а все дочери — облигатными гетерозиготными носительницами Х-сцепленного гена. Такая ситуация бывает в том случае, когда Х-сцепленное рецессивное заболевание не очень резко снижает приспособленность больного. Примером подобного заболевания является Х-сцепленный рецессивный ихтиоз или цветовая слепота к красному цвету, наследующаяся ЛГ-сцепленно.
Однако любые Лг-сцепленные рецессивные заболевания существенно чаще поражают мужчин, женщины болеют крайне редко. Это понятно, так как чтобы заболела женщи-
Рис. 5.5. vY-сцепленное рецес сивное наследование.
н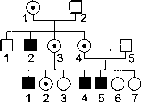 а,
должен болеть ее отец, а мать должна
быть гетерозиготой по мутантному гену.
а,
должен болеть ее отец, а мать должна
быть гетерозиготой по мутантному гену.
Для А'-сцепленных рецессивных заболеваний, как и для аутосомно-доминантных заболеваний, свойственно возникновение некоторых случаев в результате вновь возникших мутаций. Однако в отличие от аутосомно-доминантных заболеваний, когда при летальности такого заболевания на ранних этапах онтогенеза все его случаи объясняются вновь возникшими мутациями, при А-сцепленных рецессивных заболеваниях такого не бывает, поскольку у женщин, гетерозиготных носительниц летального ^-сцепленного рецессивного гена, этот ген не проявляется, так как имеется его нормальная копия во второй хромосоме X. Еще в 1935 г. В.С.Холдейн показал, что при летальности А-сцепленного рецессивного заболевания около 30 % больных «появляются» в результате вновь возникших мутаций при условии одинаковой частоты мутирования в женских и мужских половых клетках. Теперь, однако, показано, что по крайней мере большая часть мутаций в хромосоме X возникает во время сперматогенеза. В этом случае доля вновь появившихся мутаций, обусловливающих Х-сцепленное рецессивное летальное заболевание (например, миопатия Дюшенна), будет существенно меньше. Эта проблема чрезвычайно важна для медико-генетического консультирования, когда необходимо решить, является ли мать больного ребенка с А-сцепленным рецессивным летальным заболеванием гетерозиготной носительницей мутантного гена или это случай вновь возникшей мутации. В первом случае риск заболевания у ее следующего сына будет равен 50 %, а во втором — 0 %.
А-сцепленных доминантных заболеваний немного. Примером таких заболеваний являются витамин /)-резистентный гипофосфатемический рахит, при котором нарушается резорбция фосфатов в почечных канальцах и как следствие изменяется оссификация длинных трубчатых костей и происходит их искривление, что особенно характерно для нижних конечностей. Еще одно А-сцепленное доминантное заболевание — incontinentia pigmenti, проявляющееся нарушением
пигментации кожи, а также глазной и неврологической патологией и аномалиями зубов. Особенность этого заболевания в том, что все гемизиготные мужчины, носители мутантного гена, поражаются настолько сильно, что погибают внутриутробно. В результате только женщины, больные этим заболеванием, попадают в поле зрения врачей, и возникает необходимость отличать Аг-сцепленное доминантное наследование от аутосомно-доминантного наследования заболевания, ограниченного полом, такого, например, как рак молочной железы, обусловленный мутацией в гене BRCA1.
Если мутантный ген «отвечает» за Аг-сцепленное доминантное заболевание, то будут больны 50 % дочерей и 50 % сыновей больной матери, т.е., как и при аутосомно-доми- нантном заболевании, риск заболеть будет одинаковым для детей обоего пола — х/2, или 50 %. В этом случае родословная не позволяет различить эти два типа наследования. Заподозрить по родословной Аг-сцепленное доминантное заболевание позволяет родословная, в которой болен отец. Так как отец передает свою хромосому X всем дочерям, то при достаточно большом числе больных девочек и также большом числе здоровых мальчиков в семье предположение об ЛГ-сцеплен- ном доминантном заболевании, наследующемся в этой семье, может быть разумным.
Инактивация хромосомы X
Наличие у мужчин только одной хромосомы А", а у женщин двух хромосом X ставило вопрос, почему все физиологические и биохимические признаки у обоих полов проявляются примерно одинаково, хотя некоторые гены, которые контролируют эти признаки, должны быть локализованы в хромосоме X и, следовательно, должны образовывать у женщин продукта в 2 раза больше, чем у мужчин.
Цитологически уже давно было установлено, что хромосома X в соматических клетках женщин выглядит в виде плотного гетерохроматинового тельца (тельце Барра). Сначала думали, что это гетерохроматиновые районы обеих хромосом X. Однако позднее было доказано, что X-хроматин образует только одна хромосома X. Это позволяло предположить, что данная хромосома неактивна. В 1961 г. М.ФЛайон предположила, что в разных клетках у женщины может быть неактивна либо материнская, либо отцовская хромосома X. Инактивация хромосом X должна происходить достаточно рано при развитии плода, когда в эмбрионе женского пола зачатки разных органов представлены относительно небольшим числом клеток, поэтому в результате примерно 50 % клеток имеют активную отцовскую, а другие 50 % — материнскую хро-
Соматические
клехки
Рис.
5.6. Случайная
инактивация хромосомы X.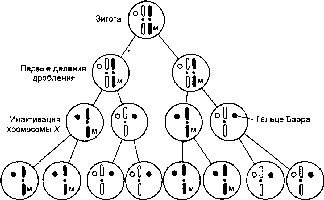
мосому X (рис. 5.6). М.Ф.Лайон также предположила, что случайная инактивация хромосомы X должна вести к мозаичному проявлению генов, локализованных в хромосоме X и находящихся в гетерозиготном состоянии, в том числе по генам, вызывающим наследственные заболевания. Это предположение было подтверждено как самой М.Ф.Лайон, так и другими исследователями, в том числе на генах ангидротиче- ской эктодермальной дисплазии, когда участки потеющей кожи перемежались с участками с отсутствием потовых желез, генах недостаточности глкжозо-6-фосфатдегидрогеназы, цветовой слепоты красно-зеленого типа и генах других сцепленных X заболеваний. Позднее было показано, что некоторые гены, особенно расположенные в теломерных концах хромосомы X, избегают инактивации. Среди этих генов довольно много таких, которые гомологичны генам хромосомы Y. Избегает инактивации также целый ряд других генов хромосомы X. ■
Частично удалось расшифровать механизм инактивации. Она начинается из определенного участка длинного плеча хромосомы X, в котором локализован центр инактивации хромосомы X. Один из генов этого центра — XIST\ мРНК, транскрибируемая с этого гена, остается в ядре и как бы покрывает хромосому X, по-видимому, инициируя таким образом начало инактивации. Во время инактивации происходит метилирование СрG-островков, расположенных в 5’-области генов, что свидетельствует об их репрессировании. Инактивация хромосомы X оказывается стабильным состоянием и сохраняется в ряду клеточных поколений. В то же время она, вероятно, не затрагивает клетки зародышевого пути или в них происходит реактивация хромосомы X, так что в зрелых яйцеклетках содержатся только активные хромосомы X.
Удивительно то, что нормальная инактивация хромосомы X, т.е. когда по 50 % клеток несет соответственно либо отцовскую, либо материнскую хромосому, обычно не приводит к каким-либо клиническим проявлениям большинства генов наследственных заболеваний, локализованных в хромосоме X, несмотря на то что их эффекты проявляются в мозаичной «манере», о чем уже было сказано. Из этого следует, что фенотипическая коррекция наследственных заболеваний возможна, когда только часть клеток поражаемого органа или ткани будет иметь нормальный генотип и соответственно фенотип.
Иногда, однако, инактивация хромосомы X происходит не случайно, и активной оказывается хромосома X, несущая мутантный ген. В этом случае у гетерозиготной женщины могут наблюдаться проявления заболевания, например миопатии Дюшенна или гемофилии (манифестирующая гетерозигота). Как правило, эти проявления заболевания бывают весьма умеренными, поскольку у таких женщин обязательно сохраняется некоторое количество клеток с нормальной хромосомой X. Инактивация одной из хромосом X в соматических клетках женщин служит для компенсации дозы гена(ов), так как она позволяет уровнять количество продуктов Х-сцеплен- ных генов у мужчин и женщин. Это подтверждается тем, что при некоторых хромосомных болезнях, когда число хромосом Xв клетках может возрасти до 4—5, активной остается только одна хромосома X, а остальные инактивируются и обнаруживаются в виде телец Барра.
Синдром ломкой хромосомы X
Этот синдром получил свое название благодаря цитогенетическим исследованиям, проведенным у мальчиков с умственной отсталостью. В клетках некоторых пациентов, культивировавшихся на среде с недостатком фолатов, наблюдались разрывы или пробелы у конца длинного плеча хромосомы X. В настоящее время известно, что, во-первых, такой феномен — ломкость хромосомы X наблюдается далеко не всегда, когда есть мутации в соответствующих генах, а во-вторых, он выявляется при наличии мутаций в любом из трех рядом расположенных генов — FRAXA, FRAXE и FRAXF.
Мы хотим изложить сведения о синдроме ломкой хромосомы X в этой главе, поскольку его наследование отличается от менделевского. Кроме того, синдром представляет собой пример динамической мутации, последнего типа мутаций, о которых шла речь в главе 4.
Клинические проявления синдрома не очень специфичны. Основным симптомом является умственная отсталость, которая у некоторых больных бывает выражена весьма умеренно. Большинство больных страдают дефицитом внимания, у некоторых наблюдают проявления аутизма. У больных старшего возраста описывают удлиненное лицо, выступающий лоб, большие оттопыренные уши и макроорхизм.
Изучение распространенности синдрома с прямым генетическим тестированием мутаций в гене FMRJ, который и обусловливает синдром, дает оценку от 16:100 ООО до 25:100 ООО мужчин; распространенность у женщин примерно в 2 раза меньше. Необычайно высока распространенность женщин с премутацией в гене FMR1 — примерно 1:300.
До недавнего времени было крайне трудно объяснить характер наследования синдрома, кодюрый проявлялся как у мужчин, так и у женщин, хотя у женщин реже и клинические симптомы у них выражены слабее. Все трудности в объяснении особенностей наследования этого синдрома удалось разрешить после того, как ген заболевания FMR1 выделили и клонировали.
Сиквенс гена показал, что в его нетранслируемой 5’-области содержится повтор ЦГГ, который у нормальных индивидуумов представлен 6—50 копиями. У больных с синдромом ломкой хромосомы X число копий повтора составляет 230 и более. Такое число называют полной мутацией. Полная мутация возникает не сразу. Сначала появляется «премутация» с числом повторов 50—230. Эта премутация может стать полной мутацией только во время созревания женских половых клеток, но не мужских. Поэтому мужчины с премутацией могут только передать ее своим дочерям. Мужчины и женщины с премутацией клинически здоровы. У женщин, носительниц премутации, во время мейоза могут происходить неожиданное увеличение числа повторов и превращение премутации в полную мутацию. Такое превращение тем более вероятно, чем больше повторов содержит премутация. Увеличение числа повторов в премутации также возникает во время созревания ооцитов у женщин-носительниц премутации с небольшим числом повторов. Полная мутация блокирует транскрипцию мРНК с гена FMR1, а повторы ЦГГ в гене оказываются метилированными. Все мужчины с полной мутацией обладают фенотипом, описанным несколькими строчками выше. У женщин с полной мутацией умственная отсталость наблюдается только в 50 % случаев, вероятно, в связи с эффектом лайонизации. Описан мозаицизм по числу повторов у лиц с полной мутацией, но остается неясным, есть ли связь между мозаицизмом и степенью проявления синдрома. Кроме того, описаны мужчины с полной мутацией, но не метилированными повторами гена FMR1, у которых не было умственной отсталости. В качестве заключения к этому разделу приведем значения риска рождения детей с полной мутацией у матерей с премутацией (табл. 5.6).
Таблица
5.6. Риск
рождения больного ребенка с полной
мутацией в гене FMR1
у
матерей с премутацией
Число
повторов ЦГГ в материнской премутации
Приблизительный
риск, %
иметь
больного сына
иметь
больную дочь
56-59
7
3,5
60-69
10
5
70-79
29
15
80-89
36
18
90-99
47
24
>
100
50
25
Примечание.
Таблица несколько видоизменена из:
Jack
Таг-
leton,
Robert A. Saul Fragile X
Syndrome
(FRAXA, Martin-Bell Syndrome, Fragile X Mental Retardation,
Marker X Syndrome) (http://www.
geneclinics.org/).