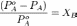1.ТЕРМОДИНАМИЧЕСКАЯ СИСТЕМА - макроскопическое тело, выделенное из окружающей среды при помощи перегородок или оболочек (они могут быть также и мысленными, условными) и характеризующееся макроскопическими параметрами: объемом, температурой, давлением и др. Для этого термодинамическая система должна состоять из достаточно большого числа частиц. 2) Однородная по химическому составу и физическим свойствам часть термодинамической системы, отделенная от др. частей (фаз), имеющих иные свойства, границами раздела, на которых происходит изменение свойств. 3)Изолированная система (замкнутая cистема) — термодинамическая система, которая не обменивается с окружающей средой ни веществом, ни энергией.
2.ЭНТАЛЬПИЯ (от греч. enthalpo - нагреваю) - однозначная функция Н состояния термодинамической системы при независимых параметрах энтропии S и давлении p, связана с внутренней энергией U соотношением Н = U + pV, где V - объем системы. При постоянном p изменение энтальпии равно количеству теплоты, подведенной к системе, поэтому энтальпию называют часто тепловой функцией или теплосодержанием. В состоянии термодинамического равновесия (при постоянных p и S) энтальпия системы минимальна. 2)Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
4.Тепловой эффект химической реакции или изменение энтальпии системы вследствие протекания химической реакции — отнесенное к изменению химической переменной количество теплоты, полученное системой, в которой прошла химическая реакция и продукты реакции приняли температуру реагентов. 2)Закон Гесса — основной закон термохимии, который формулируется следующим образом:Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы). 3)Следствия из з Гесса : Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье — Лапласа). Тепловой эффект химической реакции равен разности сумм теплот образования (ΔHf) продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты (ν): Тепловой эффект химической реакции равен разности сумм теплот сгорания (ΔHc) исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты (ν): 3)Под стандартной теплотой образования понимают тепловой эффект реакции образования одного моля вещества из простых веществ, его составляющих, находящихся в устойчивых стандартных состояниях. 4)Стандартная энтальпия сгорания — ΔHгоро, тепловой эффект реакции сгорания одного моля вещества в кислороде до образования оксидов в высшей степени окисления. Теплота сгорания негорючих веществ принимается равной нулю.
3.Первое начало термодинамики — один из трёх основных законов термодинамики, представляет собой закон сохранения энергии для термодинамических систем.
Первое начало термодинамики было сформулировано в середине XIX века в результате работ немецкого учёного Ю. Р. Майера, английского физика Дж. П. Джоуля и немецкого физика Г. Гельмгольца[1]. Согласно первому началу термодинамики, термодинамическая система может совершать работу только за счёт своей внутренней энергии или каких-либо внешних источников энергии. Первое начало термодинамики часто формулируют как невозможность существования вечного двигателя первого рода, который совершал бы работу, не черпая энергию из какого-либо источника.
5.Удельной теплоёмкостью называется теплоёмкость, отнесённая к единичному количеству вещества. Количество вещества может быть измерено в килограммах, кубических метрах и молях. В зависимости от того, к какой количественной единице относится теплоёмкость, различают массовую, объёмную и молярную теплоёмкость. 2) Чтобы рассчитать температурную зависимость энтальпии реакции, необходимо знать мольные теплоемкости веществ, участвующих в реакции. Изменение энтальпии реакции при увеличении температуры от Т1 до Т2 рассчитывают по закону Кирхгофа (предполагается, что в данном интервале температур мольные теплоемкости не зависят от температуры и нет фазовых превращений): Если в данном интервале температур происходят фазовые превращения, то при расчёте необходимо учесть теплоты соответствующих превращений, а также изменение температурной зависимости теплоемкости веществ, претерпевших такие превращения:
6.Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики гласит, что невозможен самопроизвольный переход тепла от тела, менее нагретого, к телу, более нагретому. Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая что коэффициент полезного действия не может равняться единице, поскольку для кругового процесса температура холодильника не может равняться абсолютному нулю . Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения. 2)Энтропи́я (от др.-греч. ἐντροπία - поворот, превращение) — в естественных науках мера беспорядка системы, состоящей из многих элементов. Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
7.Энтропия — функция состояния системы, равная в равновесном процессе количеству теплоты, сообщённой системе или отведённой от системы, отнесённому к термодинамической температуре системы.Энтропия — функция, устанавливающая связь между макро- и микро- состояниями; единственная функция в физике, которая показывает направленность процессов. Энтропия — функция состояния системы, которая не зависит от перехода из одного состояния в другое, а зависит только от начального и конечного положения системы. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ - один из основных законов термодинамики, закон возрастания энтропии: в замкнутой, т. е. изолированной в тепловом и механическом отношении, системе энтропия либо остается неизменной (если в системе протекают обратимые, равновесные процессы), либо возрастает (при неравновесных процессах) и в состоянии равновесия достигает максимума. Другие эквивалентные формулировки:...1) невозможен переход теплоты от тела более холодного к телу более нагретому без каких-либо других изменений в системе или окружающей среде (Р. Клаузиус);...2) невозможно создать периодически действующую (совершающую какой-либо термодинамический цикл) машину, вся деятельность которой сводилась бы к поднятию некоторого груза (механической работе) и соответственно охлаждению теплового резервуара (У. Томсон, М. Планк);...3) невозможно построить вечный двигатель 2-го рода (В. Оствальд).
8.Третье начало термодинамики (теорема Нернста) — физический принцип, определяющий поведение энтропии при приближении температуры к абсолютному нулю. Является одним из постулатов термодинамики, принимаемым на основе обобщения значительного количества экспериментальных данных. Третье начало термодинамики может быть сформулировано так:«Приращение энтропии при абсолютном нуле температуры стремится к конечному пределу, не зависящему от того, в каком равновесном состоянии находится система». Третье начало термодинамики относится только к равновесным состояниям.Поскольку на основе второго начала термодинамики энтропию можно определить только с точностью до произвольной аддитивной постоянной (то есть, определяется не сама энтропия, а только её изменение): Третье начало термодинамики позволяет находить абсолютное значение энтропии, что нельзя сделать в рамках классической термодинамики (на основе первого и второго начал термодинамики). В классической термодинамике энтропия может быть определена лишь с точностью до произвольной аддитивной постоянной , что не мешает термодинамическим исследованиям, так как реально измеряется разность энтропий в различных состояниях.
9.Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции. Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж). 2) Свобо́дная эне́ргия Гельмго́льца (или просто свобо́дная эне́ргия) — термодинамический потенциал, убыль которого в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
10.Хими́ческий потенциа́л — термодинамическая функция, применяемая при описании состояния систем с переменным числом частиц. Определяет изменение термодинамических потенциалов (энергии Гиббса, внутренней энергии, энтальпии и т. д.) при изменении числа частиц в системе. Представляет собой энергию добавления одной частицы в систему без совершения работы. dE=TdS - PdV +udN , где Е — энергия системы, S — её энтропия, N — количество частиц в системе. Эта формула определяет, кроме химического потенциала , также давление P и температуру T.
11.Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.[1] А2 + В2 = 2AB Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1885 году французским ученым Ле-Шателье. Факторы влияющие на химическое равновесие: 1) температура 2) давление 3) концентрация исходных веществ и продуктов реакции 2))Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции. для гете- К fa = П (fi в ст-vi * ak ст-vk) где fi — фугитивность компонентов газовой фазы, а ak — активность компонентов конденсированной фазы.
12.Зависимость константы равновесия реакции от температуры может быть описана уравнением изобары химической реакции (изобары Вант-Гоффа): dln Kp = дельта H / RT* x dT где Т*( в квадрате) Изобары Изохоры dln Kc = дельта U / RT* x dT
Здесь дельта Н и дельта U — тепловой эффект реакции, протекающей, соответственно, при постоянном давлении или при постоянном объёме. Если дел Н>0 (тепловой эффект положителен, реакция эндотермическая), то температурный коэффициент константы равновесия dlnKp/dT тоже положителен, то есть с ростом температуры константа равновесия эндотермической реакции увеличивается, равновесие сдвигается вправо (что вполне согласуется с принципом Ле Шателье).
13.Равнове́сие фаз в термодинамике — состояние, при котором фазы в термодинамической системе находятся в состоянии теплового, механического и химического равновесия.
Типы фазовых равновесий: 1) Тепловое равновесие означает, что все фазы вещества в системе имеют одинаковую температуру. 2) Механическое равновесие означает равенство давлений по разные стороны границы раздела соприкасающихся фаз. Строго говоря, в реальных системах эти давления равны лишь приближенно, разность давлений создается поверхностным натяжением. 3) Химическое равновесие выражается в равенстве химических потенциалов всех фаз вещества. Таким образом, равновесие фаз возможно только в том случае, когда химические потенциалы этих фаз по разные стороны границы раздела равны: u1 = u2. Фаза - Однородная по химическому составу и физическим свойствам часть термодинамической системы, отделенная от др. частей (фаз), имеющих иные свойства, границами раздела, на которых происходит изменение свойств. Потенциал Гиббса такой системы будет равен
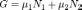 ,
,Диаграммы однокомпонентных систем
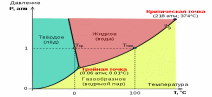

Фазовая диаграмма воды
На фазовых диаграммах однокомпонентных систем поля, по правилу фаз, соответствуют однофазным состояниям, линии, разграничивающие их — двухфазным, точки пересечения линий — трёхфазным (эти точки называют 0%A2%D1%80%D0%BE%D0%B9%D0%BD%D0%B0%D1%8F_%D1%82%D0%BE%D1%87%D0%BA%D0%B0"тройными точками).
14. Фа́зовый перехо́д (фазовое превращение) в 0%9A%D0%BB%D0%B0%D1%81%D1%81%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B0%D1%8F_%D1%82%D0%B5%D1%80%D0%BC%D0%BE%D0%B4%D0%B8%D0%BD%D0%B0%D0%BC%D0%B8%D0%BA%D0%B0"термодинамике — переход вещества из одной0%A2%D0%B5%D1%80%D0%BC%D0%BE%D0%B4%D0%B8%D0%BD%D0%B0%D0%BC%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B0%D1%8F_%D1%84%D0%B0%D0%B7%D0%B0"термодинамическойHYPERLINK "http://ru.wikipedia.org/wiki/%D0%A2%D0%B5%D1%80%D0%BC%D0%BE%D0%B4%D0%B8%D0%BD%D0%B0%D0%BC%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B0%D1%8F_%D1%84%D0%B0%D0%B7%D0%B0" фазы в другую при изменении внешних условий. С точки зрения движения системы по0%A4%D0%B0%D0%B7%D0%BE%D0%B2%D0%B0%D1%8F_%D0%B4%D0%B8%D0%B0%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D0%B0"фазовойHYPERLINK "http://ru.wikipedia.org/wiki/%D0%A4%D0%B0%D0%B7%D0%BE%D0%B2%D0%B0%D1%8F_%D0%B4%D0%B8%D0%B0%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D0%B0" диаграмме при изменении её интенсивных параметров (0%A2%D0%B5%D0%BC%D0%BF%D0%B5%D1%80%D0%B0%D1%82%D1%83%D1%80%D0%B0"температуры, 0%94%D0%B0%D0%B2%D0%BB%D0%B5%D0%BD%D0%B8%D0%B5"давления и т. п.), фазовый переход происходит, когда система пересекает линию, разделяющую две фазы. Поскольку разные термодинамические фазы описываются различными0%A3%D1%80%D0%B0%D0%B2%D0%BD%D0%B5%D0%BD%D0%B8%D0%B5_%D1%81%D0%BE%D1%81%D1%82%D0%BE%D1%8F%D0%BD%D0%B8%D1%8F"уравнениямиHYPERLINK "http://ru.wikipedia.org/wiki/%D0%A3%D1%80%D0%B0%D0%B2%D0%BD%D0%B5%D0%BD%D0%B8%D0%B5_%D1%81%D0%BE%D1%81%D1%82%D0%BE%D1%8F%D0%BD%D0%B8%D1%8F" состояния, всегда можно найти величину, которая скачкообразно меняется при фазовом переходе.
Поскольку разделение на термодинамические фазы — более мелкая классификация состояний, чем разделение по 0%90%D0%B3%D1%80%D0%B5%D0%B3%D0%B0%D1%82%D0%BD%D0%BE%D0%B5_%D1%81%D0%BE%D1%81%D1%82%D0%BE%D1%8F%D0%BD%D0%B8%D0%B5"агрегатным состояниям вещества, то далеко не каждый фазовый переход сопровождается сменой агрегатного состояния. Однако любая смена агрегатного состояния есть фазовый переход.
Наиболее часто рассматриваются фазовые переходы при изменении температуры, но при постоянном давлении (как правило равном 1 атмосфере)
Уравнение Клапейрона — Клаузиуса — термодинамическоеуравнение, относящееся к 0%9A%D0%B2%D0%B0%D0%B7%D0%B8%D1%81%D1%82%D0%B0%D1%82%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B8%D0%B9_%D0%BF%D1%80%D0%BE%D1%86%D0%B5%D1%81%D1%81"квазистатическим (равновесным) процессам0%A4%D0%B0%D0%B7%D0%BE%D0%B2%D1%8B%D0%B9_%D0%BF%D0%B5%D1%80%D0%B5%D1%85%D0%BE%D0%B4"перехода вещества из одной 0%A4%D0%B0%D0%B7%D0%B0_(%D0%B2_%D1%82%D0%B5%D1%80%D0%BC%D0%BE%D0%B4%D0%B8%D0%BD%D0%B0%D0%BC%D0%B8%D0%BA%D0%B5)"фазы в другую (испарение, плавление, сублимация, полиморфное превращение и др.). Согласно уравнению, теплота фазового перехода (например, 0%A2%D0%B5%D0%BF%D0%BB%D0%BE%D1%82%D0%B0_%D0%B8%D1%81%D0%BF%D0%B0%D1%80%D0%B5%D0%BD%D0%B8%D1%8F"теплота испарения, 0%A2%D0%B5%D0%BF%D0%BB%D0%BE%D1%82%D0%B0_%D0%BF%D0%BB%D0%B0%D0%B2%D0%BB%D0%B5%D0%BD%D0%B8%D1%8F"теплота плавления) при квазистатическом процессе определяется выражением
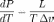
где  —
удельная теплота фазового перехода,
—
удельная теплота фазового перехода, 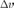 —
изменение удельного объёма тела при
фазовом переходе.
—
изменение удельного объёма тела при
фазовом переходе.
15. Фа́зовая диагра́мма воды — графическое отображение0%A2%D0%B5%D1%80%D0%BC%D0%BE%D0%B4%D0%B8%D0%BD%D0%B0%D0%BC%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%BE%D0%B5_%D1%80%D0%B0%D0%B2%D0%BD%D0%BE%D0%B2%D0%B5%D1%81%D0%B8%D0%B5"равновесного состояния фаз0%92%D0%BE%D0%B4%D0%B0"воды (0%96%D0%B8%D0%B4%D0%BA%D0%BE%D1%81%D1%82%D1%8C"жидкости, водяного параи различных модификаций0%9B%D1%91%D0%B4"льда). Строится в системе координат0%A2%D0%B5%D0%BC%D0%BF%D0%B5%D1%80%D0%B0%D1%82%D1%83%D1%80%D0%B0"температура—0%94%D0%B0%D0%B2%D0%BB%D0%B5%D0%BD%D0%B8%D0%B5"давление.
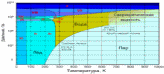
Фазовая диаграмма воды
16. Раство́р — гомогенная (однородная) 0%A1%D0%BC%D0%B5%D1%81%D1%8C_(%D1%85%D0%B8%D0%BC%D0%B8%D1%8F)"смесь, состоящая из частиц растворённого вещества, растворителя и продуктов их взаимодействия.
Раствор — однофазная система переменного состава, состоящая из двух или более компонентов. Растворы — гомогенные (однородные) системы, то есть каждый из компонентов распределён в массе другого в виде молекул, атомов или ионов0%A0%D0%B0%D1%81%D1%82%D0%B2%D0%BE%D1%80%D1%8B"[1].
Идеальным раствором называю Идеальными при любых концентрациях являются растворы, компоненты которых близки по физическим и химическим свойствам и образование которых не сопровождается объёмными и тепловыми эффектами. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными 0%A7%D0%B0%D1%81%D1%82%D0%B8%D1%86%D0%B0"частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Реальные растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области бесконечно малых 0%9A%D0%BE%D0%BD%D1%86%D0%B5%D0%BD%D1%82%D1%80%D0%B0%D1%86%D0%B8%D1%8F_%D1%80%D0%B0%D1%81%D1%82%D0%B2%D0%BE%D1%80%D0%BE%D0%B2"концентраций.
Разбавленный раствор — раствор с низким содержанием растворённого вещества. Отметим, что не всегда разбавленный раствор является 0%9D%D0%B5%D0%BD%D0%B0%D1%81%D1%8B%D1%89%D0%B5%D0%BD%D0%BD%D1%8B%D0%B9_%D1%80%D0%B0%D1%81%D1%82%D0%B2%D0%BE%D1%80"ненасыщенным — например, насыщенный 0,0000134М раствор практически нерастворимого 0%A5%D0%BB%D0%BE%D1%80%D0%B8%D0%B4_%D1%81%D0%B5%D1%80%D0%B5%D0%B1%D1%80%D0%B0"хлорида HYPERLINK "http://ru.wikipedia.org/wiki/%D0%A5%D0%BB%D0%BE%D1%80%D0%B8%D0%B4_%D1%81%D0%B5%D1%80%D0%B5%D0%B1%D1%80%D0%B0"серебраявляется очень разбавленным. Граница между разбавленным и0%9A%D0%BE%D0%BD%D1%86%D0%B5%D0%BD%D1%82%D1%80%D0%B8%D1%80%D0%BE%D0%B2%D0%B0%D0%BD%D0%BD%D1%8B%D0%B9_%D1%80%D0%B0%D1%81%D1%82%D0%B2%D0%BE%D1%80"концентрированным растворами весьма условна.
17. при замерзании раствора давление пара над твердой фазой должно быть равно давлению пара над жидкостью. Если при замерзании раствора выделяется чистый растворитель, то давление пара над жидким раствором должно быть равно давлению пара надтвердым чистым растворителем. Как было показано выше, давление пара над раствором ниже давления пара над чистым жидкимрастворителем, а следовательно, и соответствующее температуре замерзания равновесие для раствора будет устанавливаться при меньших температурах, чем для чистого растворителя. Это явление имеет важное значение в природе и технике.
Первый закон Рауля
Первый закон Рауля связывает давление 0%9D%D0%B0%D1%81%D1%8B%D1%89%D0%B5%D0%BD%D0%BD%D1%8B%D0%B9_%D0%BF%D0%B0%D1%80"насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его 0%9A%D0%BE%D0%BD%D1%86%D0%B5%D0%BD%D1%82%D1%80%D0%B0%D1%86%D0%B8%D1%8F_%D1%80%D0%B0%D1%81%D1%82%D0%B2%D0%BE%D1%80%D0%BE%D0%B2"мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
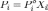
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.