

2.1.5. Метод дериватографии [41].
Общим свойством практически всех органических высокомолекулярных соединений является их термическая неустойчивость. К классу органических высокомолекулярных соединений следует относить не только регулярные полимеры (например, полиэтилен, полиакрилоншрил и др.), но и такие вещества, как пеки, угли, смолисто-асфальтеновые соединения нефти и их аналоги. При нагревании они разлагаются с выделением летучих веществ и твердого остатка.
В результате изучения процессов термического разложения было установлено, что они представляют собой совокупность физико-химических превращений и химических реакций, которые протекают в достаточно определенных температурных диапазонах.
Большое количество одновременных взаимодействий последовательно-параллельного типа и разнообразие неидентифицированных веществ, участвующих в них, делают невозможным изучение таких процессов классическими методами химической кинетики.
Практическое осуществление термодеструкции чаще всего происходит в условиях подъема температуры с постоянной или переменной скоростью, поэтому кинетика процесса в подавляющем большинстве случаев оказывается неизотермической, что тоже выдвигает свои требования к методике исследования. Удобным способом лабораторного моделирования и изучения кинетики термического разложения веществ неопределенного состава в условиях переменного температурного режима является термогравиметрия, которая основана на измерении потери массы исследуемого образца за счет выделения летучих веществ при изменении его температуры.
Сущность метода заключается в следующем: образец исследуемого вещества нагревают с постоянной скоростью до заданной конечной температуры, фиксируя при этом изменение массы. Затем при достижении образцом некоторой температурной величины начинает происходить группа реакций, скорость которых становится заметной при данной температуре. При этом скорость потери массы увеличивается и вновь падает после завершения реакций. Таких брутто-стадий при термодеструкции одного образца может быть несколько. Эти стадии могут протекать с выделением или поглощением тепла, поэтому при постоянной скорости подвода тепла рост температуры образца может ускоряться (в случае экзотермического эффекта) или замедляться (если тепловой эффект эндотермический).
Кривые потери массы и подъема температуры образца часто оказываются недостаточно информативными. В частности, по температурной кривой бывает почти невозможно определить наличие теплового эффекта реакции из-за его малой по сравнению с количеством вносимого тепла величины. Отклонение изменения температуры образца от линейности при этом практически незаметно. Иногда бывает важно знать и величину скорости химической реакции, а установить ее по кривой потери массы тоже затруднительно. Для того, чтобы устранить такие препятствия, применяют также запись скоростей потери массы и изменения температуры. Таким образом, при термогравиметрическом анализе получают кривые потери массы образца и подъема температуры, а также кривые скоростей потери массы и изменения температуры. Первые две называются интегральными, вторые — дифференциальными. Анализ, в ходе которого определяют связь потери массы образца и ее производной с изменением температуры, называют термогравиметрией, при установлении связи скорости изменения температуры с температурным режимом говорят о термографическом анализе. Методика, включающая одновременно термогравиметрический и термографический анализы, называется совмещенным термическим анализом. Приборы для его проведения называются дериватографами. Они позволяют одновременно автоматически регистрировать четыре кривые, характеризующие процесс термических превращений вещества: потерю массы (ТГ), подъем температуры образца (Т), скорость потери массы (ДТГ), которая характеризует кинетику термической деструкции, и скорость изменения температуры (ДТА), которая отражает изменение энтальпии, т.е. тепловые эффекты реакций, протекающих в массе вещества и структурно-фазовых перестроек. Все измерения осуществляются с использованием одной пробы.
Обработка полученных кривых (дериватограмм) заключается в качественном и количественном анализе кривых Т, ТГ, ДТА и ДТГ.
К изучению кинетики процессов термодеструкции по данным термогравиметрического анализа существует ряд подходов. Наиболее разработаны они для тех случаев, когда процесс проходит в одну стадию или стадии далеко разнесены по температурным интервалам и потому легко поддаются разделению на отдельные брутто-процессы. Как правило, предполагают протекание процесса по уравнению скорости реакции порядка х, подчиняющейся зависимости Аррениуса:
г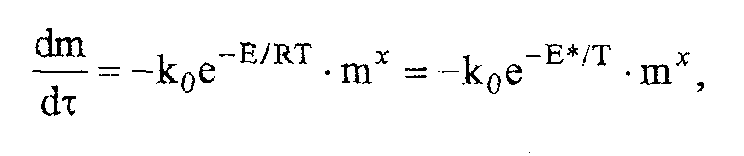 де
m
—
масса нелетучего вещества пробы в момент
времени t, Е — энергия активации, ko
—
предэкспоненциальный множитель, R
—
универсальная газовая постоянная,
Т — абсолютная температура, х
—
порядок реакции.
де
m
—
масса нелетучего вещества пробы в момент
времени t, Е — энергия активации, ko
—
предэкспоненциальный множитель, R
—
универсальная газовая постоянная,
Т — абсолютная температура, х
—
порядок реакции.
Следует заметить, что в данном случае речь должна идти не о кинетических константах вполне определенных реакций индивидуальных веществ, а о некоторых параметрах суммарных процессов термического разложения, описывающих зависимость изменения общей массы образца при повышении его температуры с определенной скоростью. По этой причине часто невозможно использовать величины ko, Е и x, определенные в одних условиях термодеструкции, для описания термического разложения того же вещества, в других условиях, например, с другой скоростью подъема температуры.
Часто для определения кинетических параметров реакции используют одну точку кривой ТГ с максимальной скоростью деструкции, и полученные результаты переносят на всю кривую в целом, что не всегда обоснованно. Тем более такой способ неприемлем при недостаточной определенности системы, включающей три неизвестных параметра. Например, сначала по степени превращения массы исходного вещества в точке перегиба оценивают порядок реакции деструкции, а затем, зная порядок, степень превращения и температуру в этой точке, определяют энергию активации и предэкспоненциалъный множитель изучаемого процесса. Полагается , что при таком подходе при расчете каждого следующего параметра ошибка его определения нарастает очень быстро. При этом, определенность системы можно повышают, принимая x=1, что для процессов деструкции часто является оправданным. Выбор первого порядка выгоден также тем, что размерность k0 включает тогда только время, и безразлично, в каких единицах измеряется количество вещества.
Более перспективны методы, использующие для определения кинетических параметров процесса весь участок кривой ТГ, относящийся к одной брутто-стадии. Этот путь позволяет заметно повысить надежность их определения, хотя и требует большой вычислительной, работы с применением ЭВМ. Далее хотелось бы рассмотреть один из таких методов.
П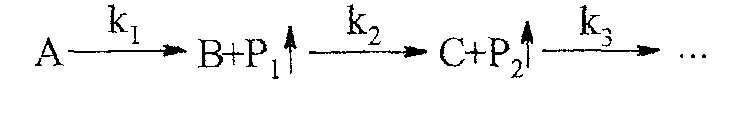 усть
исходное вещество А последовательно
разлагается, образуя промежуточный
твердый продукт В и летучий продукт Р1
.
Затем В переходит
в следующий промежуточный продукт С и
выделяет летучий продукт Р2
и т.д.:
усть
исходное вещество А последовательно
разлагается, образуя промежуточный
твердый продукт В и летучий продукт Р1
.
Затем В переходит
в следующий промежуточный продукт С и
выделяет летучий продукт Р2
и т.д.:
Скорость выделения летучих веществ на каждой стадии, как правило, можно описать уравнением реакции первого порядка с последующим определением констант скорости стадий. Процесс термической деструкции в целом будет отвечать схеме последовательных реакций первого порядка.
Допустим, что за скоростью процесса следят по общему накоплению летучих веществ, т.е. измеряют их массу за определенный промежуток времени, а число стадий деструкции равно двум (рис. 2.4.). Масса выделившихся летучих веществ при этом равна потере массы твердого образца.
Как видно из рис. 2.4, кинетическая кривая выделения летучих веществ состоит из двух стадий, и каждая из стадий представлена S-образной линией. Такая форма объясняется тем, что сначала скорость реакции растет за счет увеличения константы скорости по уравнению Аррениуса, а затем скорость деструкции падает в связи с разложением реагента.
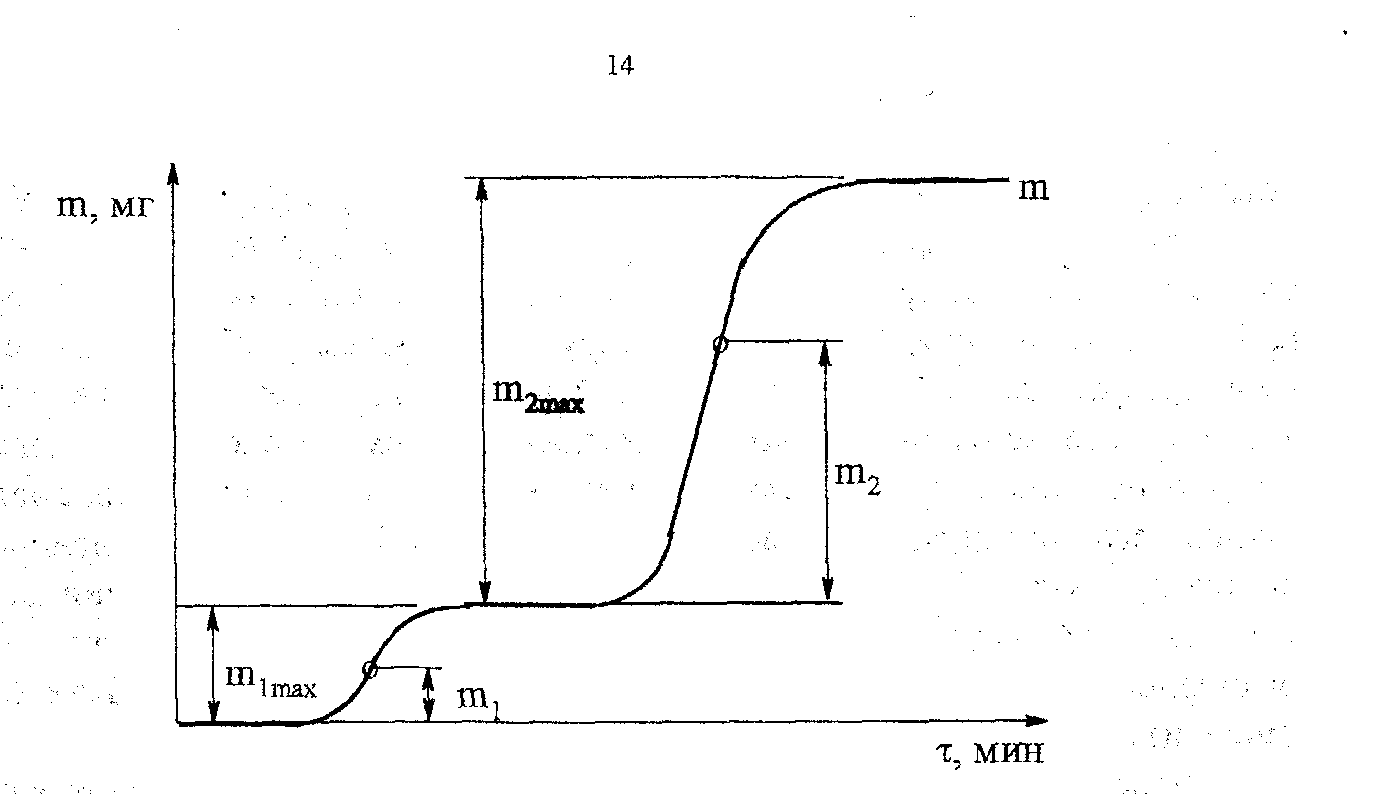
Рис. 2.4. Кривая выделения летучих веществ при термической деструкции полимера
М![]() ассы
летучих веществ, выделяющихся на каждой
стадии, различны, а число молей, участвующих
в реакции, неизвестно. В связи с этим
будем условно считать, что на каждой
стадии разлагается одно и то же число
молей
реагента, равное числу молей исходного
вещества
nA.o
.
Максимальное
число молей летучих продуктов, которое
может выделиться на каждой стадии,
также будет равно
nA.o
;
ассы
летучих веществ, выделяющихся на каждой
стадии, различны, а число молей, участвующих
в реакции, неизвестно. В связи с этим
будем условно считать, что на каждой
стадии разлагается одно и то же число
молей
реагента, равное числу молей исходного
вещества
nA.o
.
Максимальное
число молей летучих продуктов, которое
может выделиться на каждой стадии,
также будет равно
nA.o
;
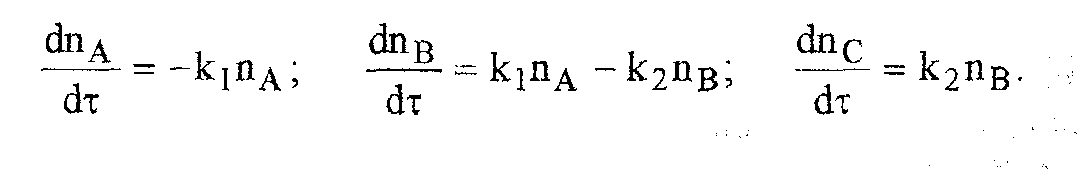 Скорость
выделения летучих веществ на некоторой
стадии разложения будет определяться
числом молей твердого исходного или
промежуточного
вещества. Тогда система уравнений,
описывающих двухстадийньй процесс
термодеструкции твердой фазы, может
быть представлена в виде:
Скорость
выделения летучих веществ на некоторой
стадии разложения будет определяться
числом молей твердого исходного или
промежуточного
вещества. Тогда система уравнений,
описывающих двухстадийньй процесс
термодеструкции твердой фазы, может
быть представлена в виде:
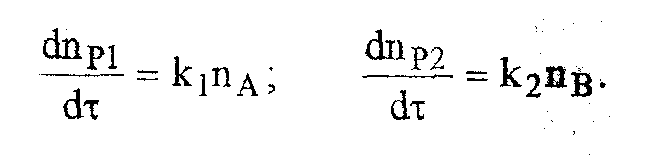 Скорости выделения
летучих веществ:
Скорости выделения
летучих веществ:
![]() Из соотношений
материального баланса:
Из соотношений
материального баланса:
с![]() другой стороны,
другой стороны,
![]() Тогда
Тогда
![]() или
или
и система уравнений будет иметь вид:
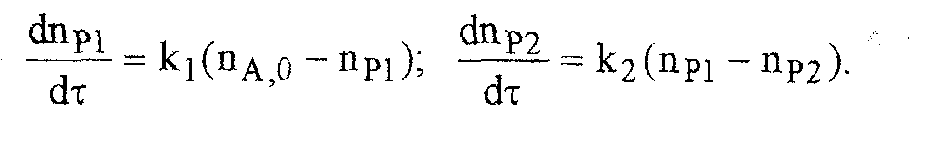
 Текущая
масса выделившихся на данной стадии
летучих веществ mi
пропорциональна
текущему числу молей npi,
(где
i
—
номер стадии кинетической
кривой выделения летучих веществ):
Текущая
масса выделившихся на данной стадии
летучих веществ mi
пропорциональна
текущему числу молей npi,
(где
i
—
номер стадии кинетической
кривой выделения летучих веществ):
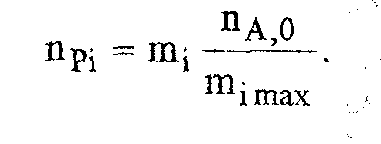
Откуда следует, что
Подставив это выражение в систему уравнений, описывающих
скорости выделения летучих веществ, получим:
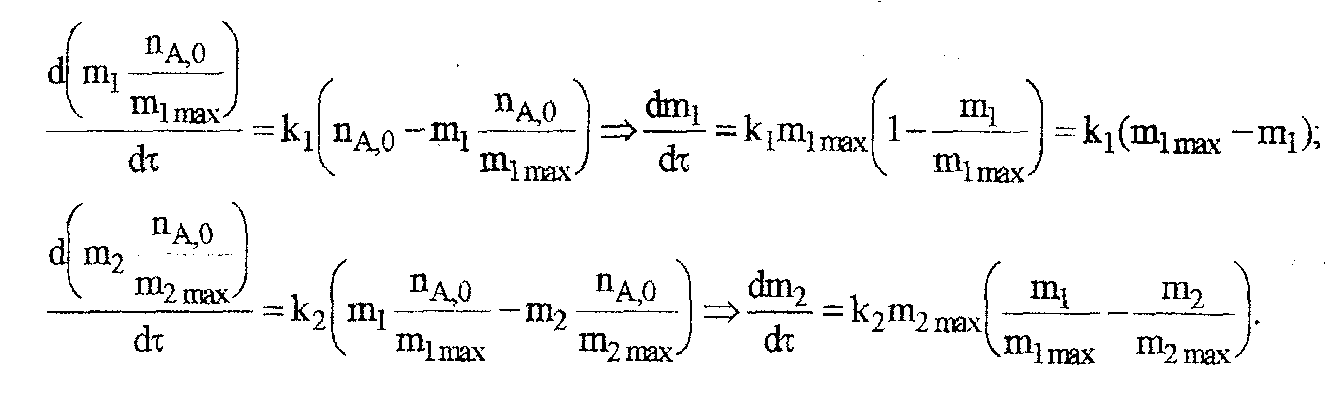
В этом случае стадии разложения далеко разнесены во .времени, поэтому к началу второй стадии выделение летучих веществ по первому уравнению уже прекратилось и m1= m1max.
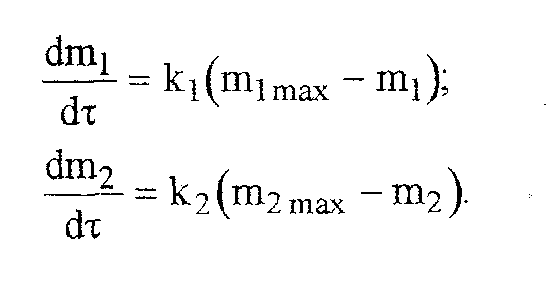 Тогда
полученная система уравнений упрощается:
Тогда
полученная система уравнений упрощается:
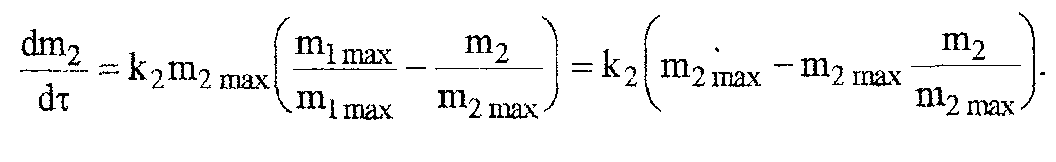 При
этом первое уравнение остается без
изменений, а второе получено
за счет преобразования
При
этом первое уравнение остается без
изменений, а второе получено
за счет преобразования
Общая потеря массы исходного вещества и скорость выделения летучих веществ:
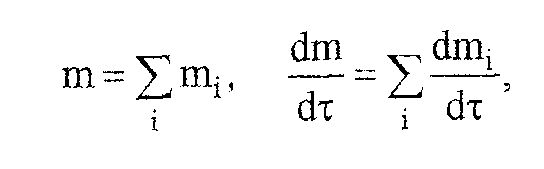
в частности, для данного случая двух стадий деструкции:
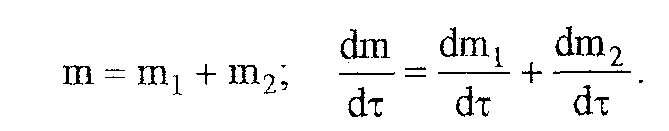
Изучаемый процесс протекает в неизотермических условиях, поэтому константы скорости k1 и k2 изменяются в ходе эксперимента в соответствии с уравнением Аррениуса. Как правило, процесс проводят таким образом, сто температура образца линейно зависит от времени реакции, не меняясь по объему образца:
г![]() де
To
–
начальная
температура,
К ;
β – скорость
подъема температуры,
де
To
–
начальная
температура,
К ;
β – скорость
подъема температуры,
K/мин.
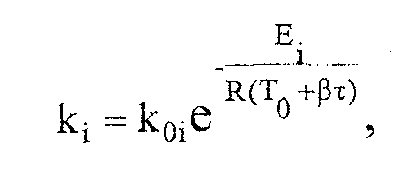 Тогда
аррениусовская зависимость констант
на i- стадии
запишется как
Тогда
аррениусовская зависимость констант
на i- стадии
запишется как
а уравнение скорости i –стадии реакции первого порядка
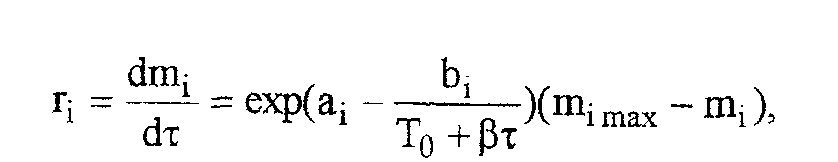
где ai= lnko ; bi= Ei/R.
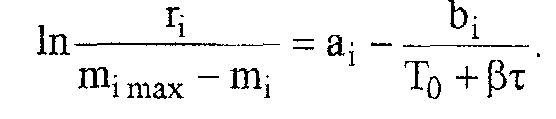 Величины
скорости изменения массы образца
определяются экспериментально как
тангенс угла наклона касательной к
кривой в заданных точках.
Подбор параметров ai=
lnko
; bi=
Ei/R
осуществляется
путем линеаризации уравнения скорости
соответствующей стадии:
Величины
скорости изменения массы образца
определяются экспериментально как
тангенс угла наклона касательной к
кривой в заданных точках.
Подбор параметров ai=
lnko
; bi=
Ei/R
осуществляется
путем линеаризации уравнения скорости
соответствующей стадии:
 Это
уравнение прямой линии в координатах
:
Это
уравнение прямой линии в координатах
:
ai –отсекаемый отрезок ординаты, bi- тангенс угла наклона.