

14.2. Электрохимическая коррозия
В коррозионной электрохимической системе идет процесс самопроизвольного разрушения металла (коррозия) при воздействии на него агрессивной среды (проводника второго рода). Процессы коррозии, подчиняющиеся закономерностям электрохимической кинетики, называются электрохимической коррозией.
Электрохимическая коррозия — сложный гетерогенный процесс, связанный с протеканием по крайней мере двух окислительно-восстановительных сопряженных реакций на поверхности корродирующего металла. В результате протекания сопряженных реакций корродирующий металл приобретает определенный стационарный потенциал, называемый в данном случае коррозионным потенциалом. Коррозионный потенциал, несмотря на необратимость процесса коррозии, может быть устойчив в течение длительного времени.
Оценка принципиальной возможности протекания электрохимической коррозии производится, как указано выше, сравнением равновесных потенциалов сопряженных электрохимических реакций. Суждение же о реальной скорости процесса коррозии можно получить лишь из кинетических зависимостей протекания сопряженных реакций. Такие зависимости представлены на рис. 14.1 для системы цинк — соляная кислота концентрации 0,1 кмоль/м3 с добавкой 10–4 кмоль/м3 хлорида цинка. Поскольку поляризация при выделении и растворении цинка невелика, катодная и анодная кривые довольно круты и ток обмена велик. Но ввиду малой концентрации ионов цинка в растворе катодная поляризационная кривая достаточно быстро выходит на предельный диффузионный ток.
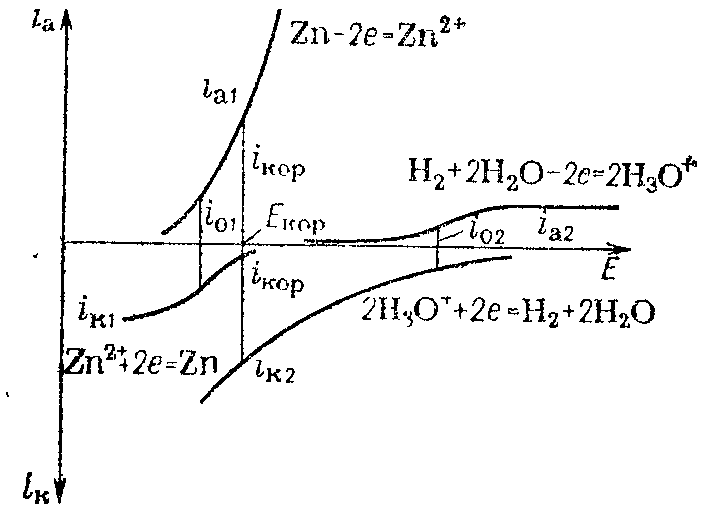
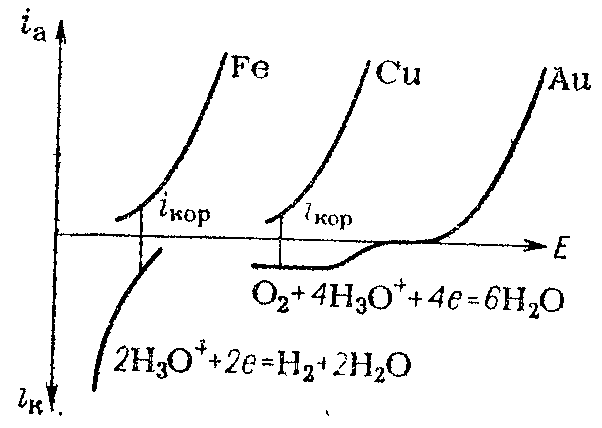
Рис. 14.1. Частные поляризационные кривые разряда — ионизации цинка и водорода на цинковом электроде.
Рис. 14.2. Расположение поляризационных кривых ионизации некоторых металлов относительно кривых восстановления ионов гидроксония и молекулярного кислорода в кислом растворе.
Перенапряжение же водорода на цинке очень велико, чем и определяются малые значения тока обмена и пологий характер поляризационной кривой. Растворение цинка и выделение водорода протекают путем передачи электронов от атома цинка ионам гидроксония, следовательно, растворение цинка будет идти со скоростью, определяемой равенством катодных и анодных токов,
т. е.
iкор = iк1 + iк2 = ia1 + ia2
Здесь индексы 1 и 2 относятся соответственно к цинку и водороду. Стационарный потенциал, отвечающий этому равенству, и будет потенциалом коррозии или коррозионным потенциалом. В рассматриваемом случае потенциал коррозии достаточно удален как от равновесного потенциала цинкового электрода, так и от равновесного потенциала водородного электрода. Вследствие этого
iк2 >> iк1; ia1 >> ia2
и можно записать
iкор = iк2 = ia1
Скорость растворения цинка является скоростью его электрохимической коррозии; она может быть охарактеризована в единицах плотности тока и называется плотностью тока коррозии. Плотность тока коррозии, а следовательно, и скорость коррозии, определяемая ординатой от Екор до пересечения с поляризационной кривой, может быть найдена не только по количеству цинка, перешедшего в раствор в единицу времени, но и по количеству выделившегося водорода, так как при коррозионном потенциале скорости этих процессов равны.
Процессом, сопряженным с растворением металла, может быть, помимо реакции восстановления ионов гидроксония, также и реакция восстановления молекул растворенного кислорода пли восстановительная реакция типа Fe3+ + e = Fe2+ и др.
Если в растворе отсутствуют вещества, способные восстанавливаться на данном металле, то процесс электрохимической коррозии не протекает, а на электроде устанавливается потенциал, связанный с адсорбцией ионов (адсорбционный потенциал). Примером такого рода потенциалов являются потенциалы, устанавливающиеся на идеально поляризуемых электродах.
На скорость коррозионного процесса и стационарный потенциал основное влияние оказывают природа металла и свойства среды. На металлах, равновесный потенциал которых положительнее равновесного потенциала водородного электрода, растворения металла не происходит и водород не выделяется. Однако если в растворе имеется растворенный кислород, который в зависимости от рН может восстанавливаться по реакции
O2 + 4Н3О+ + 4е = 6Н2O или О2 + 2Н2О + 4е = 4ОН–
то коррозия металла может протекать с сопряженной реакцией восстановления кислорода, так как равновесные потенциалы этих
реакций при одинаковом рН на 1,23 В положительнее равновесного потенциала водородного электрода (рис. 14.2).
Из расположения анодных поляризационных кривых (рис. 14.2) видно, что в слабокислых растворах, содержащих небольшие концентрации ионов соответствующих металлов, такой металл, как Fe, будет корродировать с сопряженной реакцией восстановления как ионов гидроксония, так и молекул кислорода. Но медь, равновесный потенциал которой положительнее равновесного потенциала водородного электрода в слабокислых растворах, окисляться с восстановлением ионов гидроксония не может. В присутствии растворенного кислорода сопряженной реакцией на меди будет реакция восстановления молекул кислорода. Более благородные металлы (например, Аu, а также Hg, Ag) обладают настолько высокими положительными потенциалами, что на них невозможно восстановление не только ионов гидроксония, но и растворенного кислорода, поэтому они являются устойчивыми в растворах, не содержащих других окислителей. Однако и некоторые из этих металлов (Hg, Ag) будут подвержены коррозии, если в растворе имеется окислитель с очень высоким положительным равновесным потенциалом, например азотная кислота.
Существенное влияние на коррозию оказывает рН раствора. Равновесный потенциал реакции восстановления ионов гидроксония смещается на 0,059 В в электроотрицательную сторону при повышении рН на единицу при 25 °С. Поэтому скорость коррозии металла, например цинка, будет уменьшаться при переходе от кислых к нейтральным растворам. Но в щелочных растворах цинк растворяется с образованием гидроксокомплексов по реакции
Zn + 4ОН– – 2е = Ζn(ΟΗ)42–
равновесный потенциал которой значительно отрицательнее равновесного потенциала в растворе простых ионов. Поэтому скорость коррозии цинка вновь возрастает при переходе от нейтральных к щелочным растворам.
Этот пример также показывает, какое большое влияние оказывает комплексообразование на скорость коррозии. Повышение скорости коррозии в присутствии комплексообразователя связано с ускорением процесса анодного растворения. Формально это связано со сдвигом анодной поляризационной кривой в отрицательную сторону.
Эффект комплексообразования наблюдается при коррозии золота. Золото не растворяется в азотной кислоте, так как равновесный потенциал восстановления азотной кислоты отрицательнее равновесного потенциала золота. Но золото растворяется в смеси азотной и соляной кислот (царской водке) и это связано с тем, что в этом случае золото растворяется с образованием анионов АuСl4–, а равновесный потенциал реакции
Аu + 4Сl– – 3е = АuС14–
отрицательнее равновесного потенциала восстановления азотной кислоты.
В связи с тем, что скорость реакции восстановления ионов гид- роксония определяется скоростью переноса электрона, положение поляризационной кривой этой реакции не зависит от перемешивания. Поэтому скорость коррозионных процессов с сопряженной реакцией восстановления ионов гидроксония не зависит от перемешивания раствора. Скорость же коррозии металла с сопряженной реакцией восстановления кислорода может зависеть от перемешивания, так как концентрация кислорода в растворе невелика и скорость его восстановления зачастую лимитируется стадией диффузии.
Таким образом, скорость коррозии зависит от взаимного рас-положения поляризационных кривых и механизма катодного процесса восстановления окислителя и анодного растворения металла. Значение коррозионного потенциала всегда лежит между значениями равновесных потенциалов сопряженных реакций, однако на металлах с большими плотностями тока обмена и высоким перенапряжением выделения водорода устанавливается коррозионный потенциал, весьма близкий по своему значению к равновесному потенциалу металлического электрода. Типичными примерами таких систем являются цинк в щелочном растворе цинката и амальгамы щелочных металлов в водных растворах.
При сильном же торможении скорости анодного растворения, например никеля в кислых растворах, и высоких плотностях тока обмена сопряженной реакции восстановления — ионизации водорода значение коррозионного потенциала близко к равновесному потенциалу водородного электрода при данном рН.