

13.2.2. Кинетика процесса пассивирования
Пассивирующие слои на металле не являются вполне устойчивыми по отношению к раствору. Поэтому стационарное состояние при растворении пассивного металла характеризуется параллельно протекающими реакциями образования и растворения пассивирую-щего слоя. Пассивность металла может возрастать с увеличением положительного значения потенциала, причем обычно сдвиг
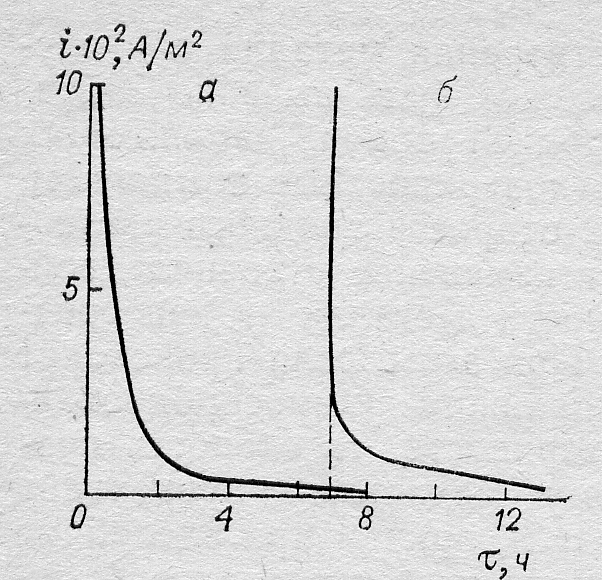
Рис. 13.2. Изменение скорости растворения хрома во времени после подачи на электрод анодного потенциала: а – Е = 0,019 В; б – Е = 0,227 В.
потенциала в положительную сторону (в область пассивности) вызывает вначале рост плотности тока, а затем ее уменьшение, иногда в течение длительного времени (рис. 13.2). Такое снижение плотности тока обусловлено двумя причинами: либо заполнением поверхности пассивирующим агентом (адсорбированным кислородом) при более положительном потенциале, либо процессом изменения состава и структуры пассивирующего слоя.
Процесс роста беспористого пассивирующего слоя на металле до толщины, большей, чем мономолекулярная, может происходить, если в нем возможна диффузия катионов к границе раздела оксид — электролит или диффузия анионов через слой оксида к поверхности металла. Другими словами, пассивирующий слой должен обладать ионной проводимостью. Движение ионов в пассивирующем слое обусловлено разностью химических потенциалов на границах и миграцией. Так как пассивирующие слои обладают сравнительно невысокой электрической проводимостью, то в них наблюдается падение потенциала, поэтому потенциал электрода в пассивном состоянии определится выражением
Е = Еп + ΔЕ
где ΔЕ — падение потенциала в слое оксида.
По мере увеличения толщины слоя при постоянной плотности тока (при постоянном ионном токе через оксид) значение ΔЕ возрастает пропорционально толщине оксида. Поэтому для таких условий должен наблюдаться линейный рост потенциала во времени. Зная толщину пленки и падение потенциала в оксиде, можно рассчитать градиент потенциала в слое, который составляет примерно 108 В/м.
Область линейной зависимости потенциала от времени пассивирования наблюдается и при образовании адсорбционных мономолекулярных слоев на благородных металлах.
Плотность ионного тока через пассивирующий оксид связана с градиентом потенциала экспоненциальной зависимостью
![]()
(где δ — толщина оксидного слоя), которая после логарифмирования имеет вид:
ln i = const + (k2/δ)ΔE
Отсюда следует линейная зависимость между логарифмом плотности ионного тока и потенциалом при постоянной толщине пленки. Экспериментальная проверка этой зависимости осуществлена на железе следующим образом. Сначала пассивируют железо в растворе серной кислоты (0,5 кмоль/м3) при потенциале 1,0 В до установления постоянной плотности тока. При этом на железе образуется пассивирующий слой толщиной δ. Затем снимают на таком электроде поляризационную кривую с достаточно быстрой разверткой потенциала от 0,8 до 1,2 В. Если за время снятия кривой не происходит изменения толщины слоя, то из предыдущих уравнений следует:
ln i = const + (k2/δ)(E – Eп)
ln i = const’ + (K2/δ)E
Зависимость плотности ионного тока от потенциала для железа в растворе H2SO4 концентрации 0,5 кмоль/м3 приведена на рис. 13.3. Аналогичная зависимость наблюдается и для запассивированного никеля.
Из последнего уравнения получается также зависимость роста толщины пленки от времени τ. Поскольку i = Q/τ, то
ln Q – 1n τ = const’ + (К2/δ)Е
или
1/δ = a – b ln τ
Это уравнение соблюдается для случаев роста пассивирующих пленок на металлах, если пассивирующие слои не растворяются в электролите (А1, Та, Ti и др.) и пассивирование проводят при постоянном потенциале и Q (рис. 13.4).
Поскольку в зависимости от состава раствора и природы металла на поверхности могут образовываться пассивирующие слои, растворяющиеся в электролите, процесс пассивирования, как указано выше, связан с соотношением скоростей образования и растворения пассивирующего слоя и, следовательно, с плотностью пассивирующего тока. Например, при анодной поляризации
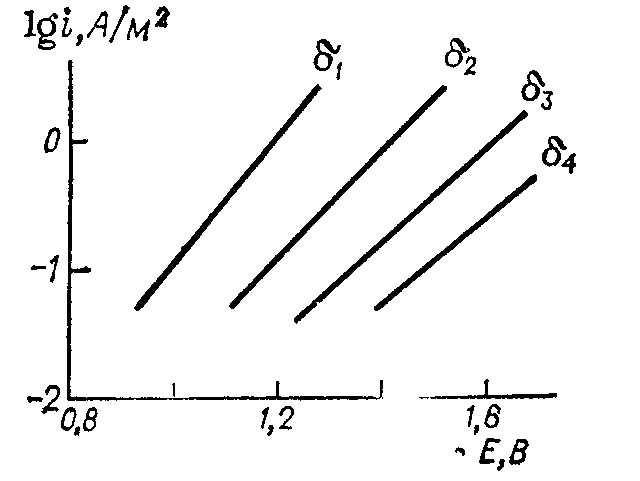
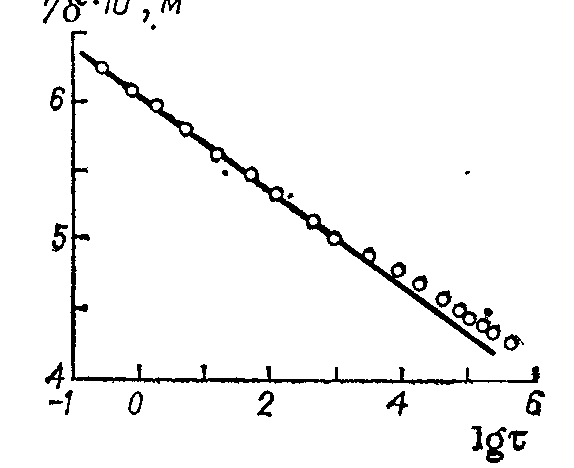
Рис. 13.3. Зависимость анодной плотности тока от потенциала пассивного железного электрода при различных толщинах оксидной пленки: δ1 < δ2 < δ3 < δ4.
Рис. 13.4. Зависимость обратной величины толщины пассивирующего слоя на тантале (Ta2O5) от времени пассивирования в потенциостатических условиях.
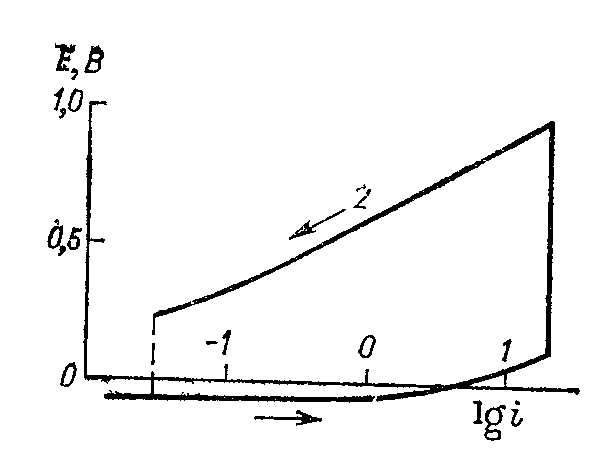
Рис. 13.5. Поляризационная кривая растворения железа в щелочи:1 — активное растворение; 2 — растворение в пассивном состоянии.
железа в растворе NaOH концентрации 10 кмоль/м3 при 80 °С и плотности тока 20 А/м2 наблюдается переход железа в пассивное состояние (рис. 13.5), но при снижении плотности тока до 3∙10–2 А/м2 железо вновь переходит в активное состояние в результате растворения в щелочи пассивирующего слоя. Явление растворения пассивирующего слоя наблюдается и при анодном растворении цинка в растворах NaOH. После перевода металла в пассивное состояние и выключения тока на нем устанавливается положительный потенциал, который с течением времени смещается в отрицательную сторону до стационарного. Время установления потенциала соответствует времени растворения пассивирующего слоя.
Пористые пассивирующие слои могут состоять и из соли металла (солевое пассивирование). В этих случаях переход электрода в активное состояние связан с концентрацией соли. Так как скорость растворения соли зависит от ее концентрации в растворе, то при высоких концентрациях соли в электролите скорость растворения пассивирующего слоя мала и пассивирование наступает при более низких плотностях тока.
Количество электричества, необходимое для образования пассивирующего слоя, который имеет способность растворяться в электролите, можно определить следующим образом. Если через электрод пропускать постоянный анодный ток, то при плотностях тока, превышающих некоторое значение, электрод переходит в пассивное состояние. Наступление пассивного состояния выражается в том, что потенциал электрода резко сдвигается в положительную сторону. Время, необходимое для наступления пассивного состояния, называется временем пассивирования τп и зависит от плотности анодного тока. Если скорость растворения пассивирующего слоя, выраженная в единицах плотности тока, равна ia’. то за время τп на растворение пассивирующего слоя расходуется ia’τп = Q1 количества электричества. Если считать, что весь ток iа идет на образование пассивирующего слоя, то за время τп должно образоваться количество, соответствующее количеству прошедшего электричества Q2 (т. е. iаτп). Очевидно, что к моменту τп на электроде осталось пассивирующего вещества, отвечающего Q2 – Q1 или
Q = Q2 – Q1 = (ia – ia’)τп
Так как ia’ соответствует скорости растворения пассивирующего слоя, которая в большинстве случаев определяется закономерностями диффузионной кинетики, то iа’ должна быть равна предельной плотности тока диффузии. Поэтому скорость реакции растворения обычно зависит от концентрации соли в растворе и перемешивания электролита. Так как для пассивирования металла обычно необходима определенная толщина пассивирующего слоя, то при различных плотностях тока должно соблюдаться равенство:
(ia – ia’)τп = const
При ia ≤ ia’ значение τп в этом равенстве должно быть либо отрицательным, либо равняться бесконечности, т. е. оно для этих условий неприменимо. Следовательно, плотность тока растворения ia’ есть одновременно и та минимальная плотность тока, при которой может произойти пассивирование. Это равенство подтверждено на примере растворения цинка в щелочи.
В случае солевого пассивирования после достижения условий его образования прохождение анодного тока и ионизация металла, сопровождаются образованием пористого пассивирующего слоя. При покрытии всей поверхности пассивирующим слоем процесс растворения перемещается в поры, в которых повышенная плотность тока вызывает пассивацию. В это время происходит растворение пассивирующего слоя в других точках. Таким образом, растворение металла осуществляется через активные мигрирующие поры. В определенных условиях растворяющиеся в электролите тонкие пассивирующие слои являются беспористыми, тогда при постоянной плотности тока наблюдаются периодические колебания потенциала.
После снятия поляризации большинство металлов через более или менее длительное время переходит в активное состояние. В ряде случаев это, как указано, связано с растворением пассивирующих слоев. Время активации зависит от толщины пассивирующего слоя и состава раствора. Активация металла катодным током, вероятно, связана с процессом восстановления оксидов либо до металла, либо до оксида низшей валентности, который растворяется в электролите. При восстановлении поверхностных оксидов катодным током рассчитывают количество электричества, пошедшее на восстановление, а если известен состав оксида, то и толщину пассивирующего слоя.
Активирующее действие на металл оказывает и анионный состав электролита. По своей активирующей способности, анионы можно расположить в ряд:
Cl– > Br– > I– > F– > ClO4– > OH–, SO42–
Активирующее действие анионов связано с адсорбционным вытеснением с поверхности электрода пассивирующего агента. Так, ионы хлора вызывают активацию железа, в щелочном растворе при положительных потенциалах, при этом происходит вытеснение адсорбированного кислорода с поверхности металла. Возможность вытеснения кислорода связана с тем, что при достаточно положительных потенциалах силы электростатического взаимодействия галогена с поверхностью настолько велики, что хлор-ионы, деформируются значительно сильнее, чем кислород.