

8.1. Поляризационные кривые
Основным отличием скорости электрохимической реакции от скорости любой гетерогенной химической реакции является ее зависимость от потенциала. Поэтому очень часто эту фундаментальную зависимость сначала выражают графически в координатах: скорость реакции в единицах плотности тока (i, А/м2) – потенциал или перенапряжение. Такая зависимость называется поляризационной кривой (катодной или анодной). Далее выводят кинетическое уравнение, описывающее данную поляризационную кривую или хотя бы часть ее и основанное на определенных представлениях о механизме протекающей реакции.
Поляризационные кривые часто имеют довольно сложный вид (рис. 8.1). При малых плотностях тока катодная и анодная поляризационные кривые имеют вид экспоненты (участок 1)*. Затем следует участок возрастающей кривой (участок 2), к которому экспоненциальное уравнение неприменимо. Участок 3 соответствует затуханию роста i с η; он переходит в площадку, отвечающую независимости плотности тока от потенциала.
На экспоненциальном участке поляризационной кривой скорость реакции обычно лимитируется стадией переноса электрона, На участке 2 скорости стадий переноса электрона и концентрационных изменений соизмеримы, в связи с чем рост плотности тока с потенциалом происходит медленнее, чем на участке 1. Участок 3 отвечает смене лимитирующей стадии. Скорость реакции здесь практически целиком ограничивается концентрационными изменениями и при концентрациях реагирующих частиц у поверхности электрода, близких к нулю, переходит в область так называемых предельных токов, не зависящих от потенциала.
В соответствии с характером поляризационных кривых эти участки имеют названия: 1) область электрохимической кинетики; 2) область смешанной кинетики; 3) область концентрационной кинетики.
Если изменение концентрации реагирующих частиц у поверхности обусловлено замедленной стадией диффузии, то область 3 называется областью диффузионной кинетики.
Кроме показанного на рис. 8.1 способа изображения поляризационных кривых, есть еще способ, представленный на рис. 8.2 и применяющийся обычно в полярографии. Согласно этому способу, вниз от линии нулевого тока откладывают анодные токи, вверх – катодные. Точка пересечения поляризационной кривой с линией
* При очень малых плотностях тока, вблизи равновесного потенциала, экспоненциальная зависимость нарушается. О причине этого см. 8.2.1.
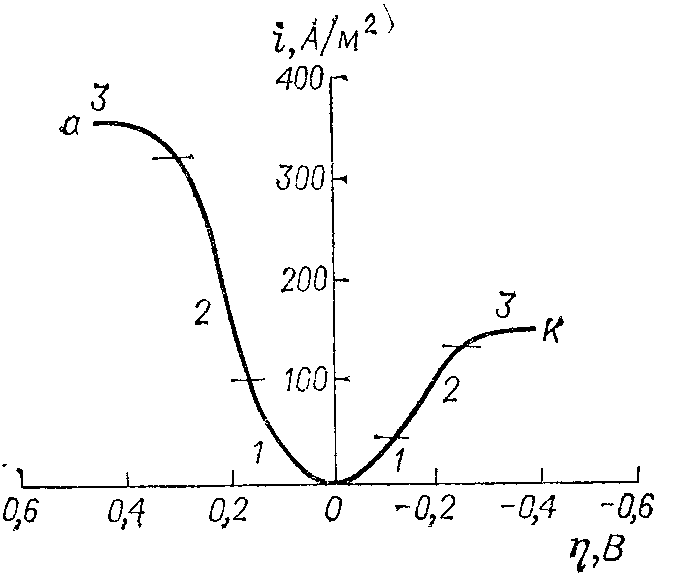

Рис. 8.1. Катодная (к) и анодная (а) поляризационные кривые на натриевом электроде в растворе NaClO4 в тетрагидрофуране концентрации 0,5 кмоль/м3.
Рис. 8.2. Поляризационные кривые реакции Ti3+ ↔ Ti4+ на ртутном капельном электроде:
1 — в растворе щавелевой кислоты (обратимый процесс); 2 — в соляной кислоте (необратимый процесс).
нулевого тока дает значение равновесного потенциала. Кривая 2 рис. 8.2 аналогична кривым рис. 8.1 и соответствует необратимому процессу, кривая 1 – обратимому.
В случае замедленного протекания электрохимической стадии происходит разделение волны на анодную и катодную и такие кривые соответствуют необратимому процессу. Если же скорость процесса лимитируется стадией диффузии и поляризационная кривая представляет собой единую анодно-катодную волну, то такой процесс называется обратимым. В этом случае перенапряжение определяется сдвигом равновесного (обратимого) потенциала из-за изменения концентрации потенциалопределяющих ионов у поверхности электрода. При изменении условий эксперимента может измениться природа замедленной стадии, а следовательно, и обратимость процесса. Кроме того, обратимость или необратимость процесса в определенной мере связана с точностью эксперимента. Так, при увеличении точности эксперимента процесс, казавшийся обратимым, может проявить характерные черты необратимости.
Методы экспериментального получения стационарных поляризационных кривых разнообразны. В гальвано- и потенциостатическом методах поддерживаются постоянными либо плотность тока, либо потенциал, а другой параметр (соответственно, потенциал или ток) регистрируется, когда достигает стационарного значения. Полученные таким методом кривые, называются стационарными поляризационными кривыми.
В динамических методах ток или потенциал изменяются с течением времени по определенному закону (обычно линейно) и регистрируется потенциал или ток, соответственно, как функция первого параметра. Такие поляризационные кривые называются динамическими, При снятий динамических кривых скорость

Рис. 8.3. Схема, поясняющая построение поляризационной кривой при задании постоянного потенциала (а) и постоянного тока (б).
изменения (развертки) тока или потенциала может изменяться в очень широких пределах – от сотых долей милливольта в секунду до сотен вольт в секунду. Если скорости развертки достаточно малы, то динамические поляризационные кривые, как правило, совпадают со стационарными и могут быть интерпретированы как стационарные поляризационные кривые. При высоких скоростях развертки динамические кривые существенно отличаются от стационарных, что связано как с протеканием процесса в нестационарных условиях, так и с наложением дополнительных факторов, например нефарадеевских токов (токов заряжения).
Поляризационные кривые сложных процессов существенно зависят от метода снятия кривой (с заданием тока или потенциала). Например, для кривой, изображенной на рис. 8.3, а любому заданному значению потенциала Е1 – Е4 соответствует вполне определенное значение тока i1 – i4. Если поддерживать постоянным ток, то, как видно из рис. 8.3, б, току i1 соответствует только потенциал Е1, току i2 – потенциал Е2 или область потенциалов E3 – E4, току i3 – потенциал Е5 и Е6, току i4 – потенциал Е7. Экспериментально это выражается в том, что при определенном значении тока i потенциал в области E2 – Е4 «ползает», а кривая, полученная с заданием тока, имеет вид, изображенный на рис. 8.4.

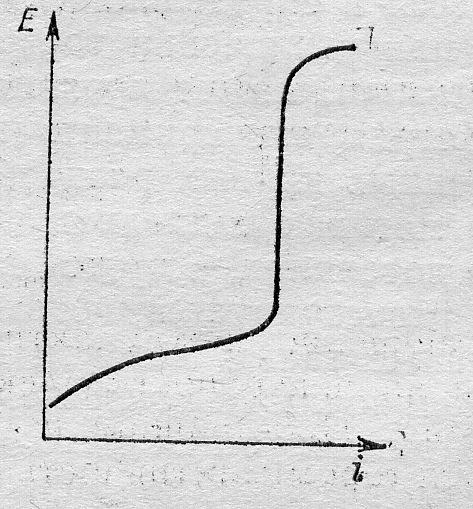
Рис. 8.4. Поляризационная кривая (рис. 8.3, б), полученная при задании постоянного тока.
Рис. 8.5. Правильное изображение поляризационной кривой, полученной в гальваностатическом режиме.
Строго говоря, при графическом изображении потенцио- и гальваностатических кривых координатные оси должны быть различны. Поскольку по оси х откладывается аргумент, а по оси y – функция, то кривые, изображенные на рис. 8.3, а и б, соответствуют потенциостатическому режиму, а гальваностатическому режиму отвечает кривая на рис. 8.5.
В литературе по электрохимии такое различие в построении поляризационных кривых не является общепринятым, поэтому следует указывать, каким способом получена та или иная кривая.%)%