

Vnutrennie_bolezni_6-e_izd_Makolkin
.pdfСтратификация риска
Стратификация риска базируется в основном на наличии в анамнезе синкопальных состояний, эпизодов полиморфной желудочковой тахикардии типа torsades de pointes и остановок кровообращения.
Лечение
Основная задача лечебных мероприятий - профилактика внезапной сердечной смерти.
Все больные с синдромом удлиненного интервала Q-T должны ограничивать физические нагрузки, в особенности избегать спортивных соревнований. Это положение обязательно для всех больных, имеющих вариант LQTS1. Для лиц, имеющих вариант LQTS3, с определенной осторожностью возможно выполнение некоторых физических нагрузок, исключающих спортивные соревнования.
Крайне важно информировать больных с синдромом удлиненного интервала Q-T, что они должны избегать приема лекарств, способных вызвать удлинение периода Q-T (многие антибиотики, антидепрессанты и др.).
В качестве медикаментозной терапии профилактики внезапной сердечной смерти могут использоваться бета-адреноблокаторы. Однако они не способны обеспечить полную защиту и для больных, имевших в анамнезе остановку кровообращения, риск внезапной сердечной смерти остается очень высоким.
Именно поэтому тем больным, у которых, несмотря на прием адекватной дозы бетаадреноблокаторов, продолжают возникать обморочные состояния, может выполняться левосторонная симпатэктомия, приводящая к симпатической денервации сердца.
Однако наиболее эффективным способом предупреждения внезапной сердечной смерти у больных с синдромом удлиненного интервала Q-T является имплантация кардиовертеров-дефибрилляторов. Их применение рекомендуется у лиц, перенесших остановку кровообращения, также у детей при наличии синдрома Джеруэлла-Ланге- Нилсена, сопровождающегося клиническими проявлениями заболевания.
Профилактика
Профилактики возникновения синдрома длинного интервала Q-T не существует.
Синдром Бругада
Синдром Бругада - вариант ионной каналопатии, описанный братьями Brugada в 1992 г., относящийся к первичным генетически обусловленным кардиомиопатиям («электрические болезни миокарда»), при которых существует высокая вероятность внезапной сердечной смерти.
В основе заболевания лежит мутация, приводящая к нарушению функции а- субъединицы №+-каналов кардиомиоцитов, кодируемых геном SCN5 (этот же ген ответствен за возникновение синдрома LQTS3). Сообщается также и о мутации в хромосоме 3, однако конкретный ген пока еще не идентифицирован.
281
Семейные формы синдрома Бругада передаются по аутосомно-доминантному типу наследования.
Клиническая картина
Клинически синдром Бругада характеризуется возникновением синкопальных и пресинкопальных состояний, также развитием внезапной сердечной смерти. Наиболее часто последняя наблюдается у молодых мужчин возрасте 30-40 лет, проживающих в Юго-Восточной Азии (Япония, Тайланд, Филиппины, Камбоджа). Частота остановки кровообращения в течение 3 лет составляет 30% как у лиц с клиническими проявлениями заболевания, так и без них. Причиной внезапной сердечной смерти у больных с синдромом Бругада являются полиморфные желудочковые тахикардии с высокой частотой возбуждения желудочков сердца, возникающие в покое либо во время сна (рис.
2-5).
Диагностика
Диагностика синдрома Бругада основана на характерных изменениях ЭКГ, проявляющихся возникновением преходящей блокады правой ножки пучка Гиса и подъемом сегмента S-T на ЭКГ в отведениях V1-V3 (рис. 2-6).
Лечение
Лечение больных с синдромом Бругада сводится к профилактике внезапной сердечной смерти. К сожалению, ни один из лекарственных препаратов не оказался эффективным для предупреждения внезапной сердечной смерти у больных с синдромом Бругада. Наилучшим методом профилактики внезапной смерти у больных с синдромом Бругада, имевших в анамнезе обморочные состояния либо остановку кровообращения, а также больных без клинических проявлений заболевания, служит имплантация кардиовертерадефибриллятора.
Профилактика
Профилактики возникновения синдрома Бругада не существует.
282
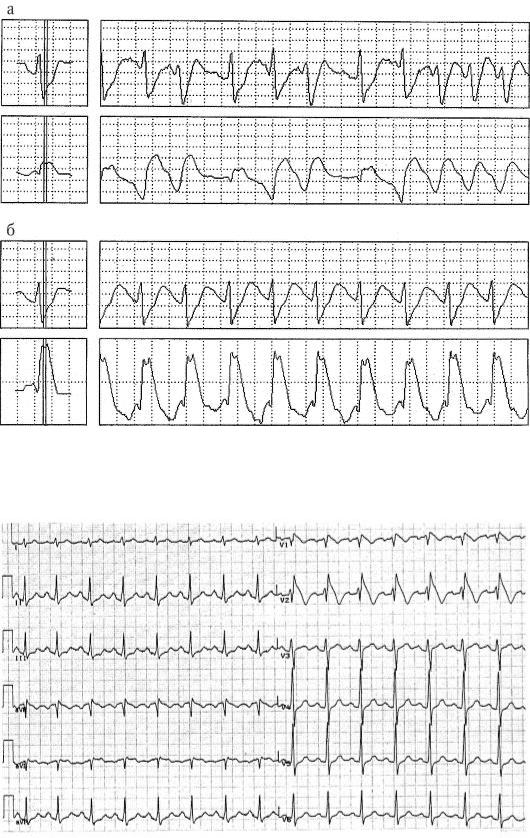
Рис. 2-5. Фрагмент холтеровского мониторирования ЭКГ во время возникновения эпизода полиморфной (а) и мономорфной (б) желудочковой тахикардии у больного с синдромом Бругада
Рис. 2-6. ЭКГ больного с синдромом Бругада
283
Катехоламинергическая полиморфная желудочковая тахикардия
Катехоламинергическая полиморфная желудочковая тахикардия (КПЖТ) - одна из форм ионных каналопатий, относящихся к первичными генетически обусловленным кардиомиопатиям («электрические болезни миокарда).
Причиной возникновения КПЖТ считают возникновение мутации в гене рианодиновых рецепторов сердца человека (hRyR2), расположенном на хромосоме lq42-q43. Рианодиновые рецепторы hRyR2 - ключевой белок, регулирующий высвобождение Са2+ из саркоплазматического ретикулума и сопряжение процессов возбуждения и сокращения в кардиомиоцитах. Заболевание передается по аутосомно-доминантному принципу наследования.
Клиническая картина
Клиническая картина КПЖТ проявляется возникновением обмороков либо предобморочных состояний, выраженных головокружений на фоне приступов сердцебиений. Однако наиболее серьезным клиническим проявлением КПЖТ служит развитие внезапной сердечной смерти. Для больных с КПЖТ характерно возникновение желудочковой аритмии под влиянием адренергической стимуляции при отсутствии какихлибо структурных изменений сердца. Пациенты, страдающие КПЖТ, чаще всего оказываются на приеме у кардиолога в связи с развитием у них синкопальных состояний, при этом примерно у 30% пациентов в семейном анамнезе отмечаются случаи обмороков
ивнезапной сердечной смерти.
Диагностика
ЭКГ в 12 отведениях, зарегистрированная в покое, может быть неизмененной, за исключением синусовой брадикардии и выраженной волны U у некоторых больных. Во время приступа аритмии на ЭКГ регистрируется картина, свойственная полиморфной желудочковой тахикардии, для которой характерно наличие тахикардии с широкими комплексами QRS и высокой частотой возбуждений желудочков, а также чередование направленности комплексов QRS (рис. 2-7).
284

Рис. 2-7. Полиморфная желудочковая тахикардия
Синдром короткого интервала Q-T
Синдром короткого интервала Q-T (SQTS) - представляет собой одну из форм ионных каналопатий, относящихся к первичным генетически обусловленным кардиомиопатиям («электрические болезни миокарда»). Этот синдром был описан недавно, в 2000 г., и характеризуется высокой вероятностью внезапной сердечной смерти вследствие жизнеугрожающих желудочковых тахиаритмий (желудочковая тахикардия, фибрилляция желудочков) у лиц без какой-либо органической патологии сердца. Синдром короткого интервала Q-T обусловлен мутациями в генах, контролирующих входящие внутрь клеток калиевые токи IK, Iks, Ikl. В основе синдрома SQT1 лежит мутация в гене KCNH2, синдром SQT2 обусловлен мутацией гена KCNQ1, синдром SQT3 связан с мутацией в гене
KCNJ2.
Клиническая картина
Клиническая картина синдрома короткого интервала Q-T складывается из обморочных состояний и развития внезапной сердечной смерти (вследствие внезапно возникающих желудочковых тахиаритмий) при отсутствии какоголибо органического заболевания сердца.
Диагностика
Диагностика синдрома короткого интервала Q-T основана на ЭКГпризнаках, основными из которых служат укорочение корригированного интервала Q-T менее 330 мс (0,33 с), а также выявление высоких заостренных зубцов Т, схожих по морфологии с зубцами Т, регистрирующимися при гиперкалиемии .
285
Лечение
Лечение больных с синдромом короткого интервала Q-T, у которых возникали синкопальные состояния либо были зарегистрированы эпизоды желудочковых тахиаримтий, сводится к имплантации кардиовертера-дефибриллятора. Медикаментозных способов предупреждения внезапной сердечной (аритмической) смерти у этой категории больных на сегодняшний день не существует.
Профилактика
Профилактики возникновения синдрома короткого интервала Q-T не существует.
ДИЛАТАЦИОННАЯ КАРДИОМИОПАТИЯ
Дилатационная кардиомиопатия (ДКМП) относится к первичным кардиомиопатиям, имеющим смешанное происхождение, т.е. она может быть как генетически обусловленной, так и развиваться под влиянием других, негенетических факторов.
ДКМП характеризуется выраженным расширением камер сердца, преимущественно левого и правого желудочков, в сочетании со снижением их сократительной функции и отсутствием гипертрофии миокарда.
Заболеваемость составляет 1 случай на 2500 человек, при этом болезнь в 2-3 раза чаще встречается у мужчин среднего возраста (30-50 лет).
С течением времени ДКМП приводит к снижению сократительной функции левого и правого желудочков, развитию прогрессирующей сердечной недостаточности, возникновению наджелудочковых (МА) и желудочковых нарушений ритма, нарушений проводимости сердца, возникновению тромбов в полостях сердца с развитием тромбоэмболических осложнений и в конечном итоге - к смерти либо вследствие фатальных желудочковых аритмий (внезапная сердечная смерть), либо вследствие насосной дисфункции сердца.
Примерно у 30% больных отмечена семейная (генетически обусловленная) форма заболевания, при которой преобладает аутосомно-доминантный тип наследования, реже встречаются аутосомно-рецессивные и Х-сцепленные формы ДКМП. Некоторые мутации, приводящие к развитию ДКМП касаются генов, кодирующих синтез таких сократительных белков, как кардиальный а-актин, а-тропомиозин, сердечные тропонины Т и I, тяжелые цепи а- и бета-миозина, миозинсвязывающий протеин С и др. При Х- сцепленных формах ДКМП выявляют мутации генов, ответственных за синтез дистрофина, десмина, кавеолина, а- и бета-саркогликана.
Существуют и другие причиной развития ДКМП: вирусные инфекции, нередко вызывающие миокардиты (вирусы Коксаки, гепатита С, герпеса, цитомегаловирусы, аденовирусы, парвовирусы, ВИЧ), бактериальные инфекции, риккетсии, микобактерии и паразиты (например болезнь Шагаса, возникающая вследствие инфицирования
Trypanosoma cruzi).
Помимо этого к причинам, вызывающим ДКМП, следует отнести ряд интоксикаций: чрезмерное употребление алкоголя (алкогольная форма ДКМП), интоксикации рядом
286
металлов (кобальт, свинец, ртуть, мышьяк), применение химиотерапевтических препаратов (доксорубицин и др.).
ДКМП может развиваться у больных аутоиммунными заболеваниями, в том числе при системных заболеваниях соединительной ткани. Однако примерно в половине случаев этиология ДКМП остается неизвестной (идиопатическая форма).
Патогенез
В результате воздействия этиологических факторов возникает повреждение кардиомиоцитов с уменьшением количества нормально функционирующих мышечных элементов. Это приводит к возникновению и прогрессированию сердечной недостаточности, проявляющейся в значительном снижении сократительной способности миокарда, т.е. развивается систолическая дисфункция желудочков сердца с быстрым развитием дилатации всех его камер.
Прогрессирование патологического процесса приводит к критическому снижению насосной функции сердца, повышению конечно-диастолического давления в желудочках, развитию миогенной дилатации полостей сердца с формированием относительной недостаточности митрального и трехстворчатого клапанов. Одновременно происходящая активация нейрогормональных систем регуляции кровообращения (симпатоадреналовой, РААС) приводит к еще большему повреждению сохранившихся кардиомиоцитов (см. раздел «Хроническая сердечная недостаточность»), снижению ударного и минутного объемов сердца, повышению периферического сопротивления и к развитию застойной сердечной недостаточности в малом, а в дальнейшем и в большом круге кровообращения.
Нарушается активность свертывающей и противосвертывающей систем крови с развитием внутрисердечных тромбов и последующих системных тромбоэмболий.
Клиническая картина
Заболевание чаще возникает у лиц молодого и среднего возраста. Специфических признаков заболевания нет.
Клиническая картина полиморфна и складывается из:
•симптомов хронической сердечной недостаточности;
•нарушений ритма и проводимости сердца;
•тромбоэмболических осложнений.
Все эти явления развиваются по мере прогрессирования заболевания, поэтому распознавание ДКМП на ранних стадиях представляет значительные трудности. На ранних этапах выявляют лишь начальные признаки сердечной недостаточности (одышку при выраженных физических нагрузках, снижение работоспособности, повышенную утомляемость).
На первом этапе диагностического поиска в ранней стадии заболевания симптомы могут не выявляться. При снижении сократительной функции миокарда появляются жалобы на повышенную утомляемость, одышку при физической нагрузке, а затем в покое. По ночам беспокоит сухой кашель (эквивалент сердечной астмы), позже - типичные
287
приступы удушья. У 10% больных наблюдают характерные ангинозные боли. При развитии застойных явлений в большом круге кровообращения появляются тяжесть в правом подреберье (вследствие увеличения печени), отеки ног.
На втором этапе диагностического поиска наиболее важный признак - значительное увеличение сердца (признаки клапанного порока сердца или АГ отсутствуют). Обнаружение на ранних стадиях болезни кардиомегалии в большей или меньшей степени может быть случайным во время профилактического осмотра или обращения больного к врачу по поводу сердечных жалоб. Кардиомегалия проявляется расширением сердца в обе стороны, определяемым перкуторно, а также смещением верхушечного толчка влево и вниз. В тяжелых случаях при аускультации сердца I тон на верхушке ослаблен, выслушиваются ритм галопа, тахикардия, шумы относительной недостаточности митрального и (или) трехстворчатого клапанов. В 20% случаев развивается МА. АД обычно нормальное или слегка повышено (вследствие сердечной недостаточности).
Остальные симптомы появляются только при развитии сердечной недостаточности и служат ее выражением (холодные цианотичные конечности, набухание шейных вен, отеки, увеличение печени, застойные хрипы в нижних отделах легких, увеличение числа дыханий в минуту).
На третьем этапе диагностического поиска (при отсутствии признаков почечно-
печеночной недостаточности при далеко зашедших стадиях хронической сердечной недостаточности) лабораторно никаких изменений выявить не удается.
Инструментальные методы исследования позволяют обнаружить:
•рентгенологические признаки кардиомегалии и застоя в малом круге кровообращения;
•на ЭКГ могут регистрироваться нарушения ритма и проводимости;
•основная диагностически значимая информация может быть получена с помощью ультразвукового исследования сердца.
•ЭхоКГ оказывает существенную помощь в диагностике, выявляя:
-дилатацию желудочков со снижением сердечного выброса;
-снижение движения стенок желудочков;
-парадоксальное движение межжелудочковой перегородки во время систолы;
-в допплеровском режиме можно обнаружить относительную недостаточность митрального и трехстворчатого клапанов.
•Ультразвуковыми диагностическими критериями ДКМП считают снижение фракции выброса левого желудочка менее 45% и увеличение конечнодиастолического размера левого желудочка более 117% от надлежащего (рассчитывают на основании возраста и площади поверхности тела больного).
•Рентгенологически обнаруживают значительное увеличение желудочков (часто в сочетании с умеренным увеличением левого предсердия). Развивающиеся вследствие левожелудочковой недостаточности нарушения в малом круге кровообращения
288
проявляются усилением легочного сосудистого рисунка, а также появлением транссудата
вплевральных (чаще в правой) полостях.
•На ЭКГ не отмечают каких-либо характерных изменений или сдвиги носят неспецифический характер. К ним относят признаки гипертрофии левого желудочка и левого предсердия; нарушения проводимости в виде блокады передней ветви левой ножки предсердно-желудочкового пучка (пучка Гиса) или полной блокады левой ножки (15% случаев); уплощение зубца Т в левых грудных отведениях; мерцание предсердий. Некоторые сложности возникают при появлении патологических зубцов Q в грудных отведениях, что заставляет заподозрить перенесенный ранее ИМ. При морфологическом исследовании миокарда в таких случаях обнаруживают множество мелких рубчиков (не являющихся следствием коронарного атеросклероза).
•ФКГ подтверждает аускультативные данные в виде ритма галопа, довольно частого обнаружения систолического шума (вследствие относительной недостаточности митрального или трехстворчатого клапана). При застойных явлениях в малом круге кровообращения выявляют акцент II тона.
•Дополнительные инструментальные исследования не являются обязательными для постановки диагноза, однако их результаты позволяют детализировать степень расстройств гемодинамики и характер морфологических изменений миокарда.
•Исследование показателей центральной гемодинамики выявляет низкий минутный и ударный объем (минутный и ударный индексы), повышение давления в легочной артерии.
•Ангиокардиографически обнаруживают те же изменения, что и на ЭхоКГ.
•Биопсия миокарда позволяет проводить дифференциальный диагноз ДКМП со вторичными кардиомиопатиями, протекающими с выраженной кардиомегалией. При вторичных кардиомиопатиях в биоптатах можно обнаружить признаки миокардита, васкулита, болезней накопления (гемохроматоза, амилоидоза) и др.:
-при тяжелых диффузных миокардитах обнаруживается клеточная инфильтрация стромы в сочетании с дистрофическими и некротическими изменениями кардиомиоцитов;
-при первичном амилоидозе, протекающем с поражением сердца (так называемый кардиопатический вариант первичного амилоидоза), наблюдается значительное отложение амилоида в интерстициальной ткани миокарда, сочетающееся с атрофией мышечных волокон;
-при гемохроматозе (заболевание, обусловленное нарушением обмена железа) в миокарде находят отложения железосодержащего пигмента, наблюдают различной степени дистрофию и атрофию мышечных волокон, разрастание соединительной ткани.
Диагностика
Ключевой момент в диагностике ДКМП - исключение других причин развитиях сердечной недостаточности.
Распознавание ДКМП представляет существенные трудности, так как значительное увеличение сердца с отсутствием или наличием сердечной недостаточности встречается с большей или меньшей частотой при других заболеваниях сердца. Среди этих заболеваний
289
- диффузные миокардиты тяжелого течения, ИБС (постинфарктный кардиосклероз с развитием аневризмы сердца), приобретенные пороки сердца в стадии тотальной сердечной недостаточности, ГБ далеко зашедших стадий, болезни накопления (гемохроматоз, первичный амилоидоз с преимущественным поражением сердца).
Лечение
Основное лечение ДКПМ - борьба с развивающейся застойной сердечной недостаточностью, что осуществляется по общим принципам (см. раздел «Хроническая сердечная недостаточность»).
Прежде всего, это касается общих мероприятий, касающихся ограничения физической активности, употребляемого количества жидкости до 1,5 л и поваренной соли до 1-2 г/сут.
Высокоэффективны ингибиторы АПФ: каптоприл в дозе 25-100 мг/сут, эналаприл 2,5-20 мг/сут, рамиприл 1,25-10 мг/сут, периндоприл по 4-8 мг/сут, лизиноприл 10-20 мг/сут. При назначении этих препаратов следует учитывать величину АД, так как они снижают его.
Для того чтобы избежать возникновения гипотонии, лечение начинают с небольших доз и, убедившись в отсутствии выраженного гипотензивного эффекта, дозу препарата увеличивают.
В комплексную медикаментозную терапию хронической сердечной недостаточности целесообразно включать (при их переносимости) бета-адреноблокаторы. С этой целью могут использоваться как селективные бета-адреноблокаторы (метопролол, бисопролол, небиволол), так и неселективные (карведилол). При этом начальная доза бетаадреноблокатора должна быть очень низкой с последующим постепенным ее увеличение до достижения максимально переносимой либо так называемой «целевой» дозы (см. раздел «Хроническая сердечная недостаточность»).
Больным с признаками задержки жидкости в огранизме (застой в легких, периферические отеки, гепатомегалия, асцит, анасарка) должны назначать мо-
чегонные препараты. Преимущество отдают петлевым диуретикам - фуросемиду, торасемиду. Доза препарата и частота приема колеблются в зависимости от стадии недостаточности кровообращения. Рекомендуют начинать лечение с небольших доз: фуросемид 20-40 мг утром натощак 1-3 раза в неделю.
Дигоксин назначают при сочетании клинической картины выраженной сердечной недостаточности (II-IV класс NYHA) и МА в обычных дозах (при этом следует иметь в виду, что при ДКМП может быстро развиться дигиталисная интоксикация, поэтому контроль приема препарата должен быть строгим).
При ДКМП можно проводить трансплантацию сердца. Основные показания - тяжелая застойная сердечная недостаточность и отсутствие эффекта от лекарственной терапии.
РЕСТРИКТИВНАЯ КАРДИОМИОПАТИЯ
Рестриктивная кардиомиопатия (РКМП) - это редкая форма заболевания сердца, которая приводит к развитию хронической сердечной недостаточности и характеризуется уменьшенным (либо нормальным) объемом обоих желудочков, неизмененным клапанным
290